miR-24對氧化低密度脂蛋白誘導血管內皮細胞炎癥損傷的影響
2022-02-17 04:01:30趙燕姣楊鵬羅雪蘭周福隆歐和生
中國老年學雜志 2022年2期
趙燕姣 楊鵬 羅雪蘭 周福隆 歐和生
(1廣西醫科大學藥學院,廣西 南寧 530201;2廣西中醫藥大學附屬國際壯醫醫院科技部)
動脈粥樣硬化(AS)是一種慢性炎性疾病,是導致血管疾病死亡的主要原因之一。血管內皮細胞(VEC)功能紊亂和炎癥損傷是驅動AS斑塊形成、發展和破裂的關鍵細胞事件。氧化低密度脂蛋白(Ox-LDL)是一種促AS發展的關鍵因子,其通過與LOX-1結合,促進促炎細胞因子的釋放,細胞黏附分子(ICAM)、單核細胞趨化蛋白-1和平滑肌細胞生長因子的過表達,導致VECs功能紊亂和炎癥反應的發生〔1〕。然而,內皮細胞炎癥反應過程中涉及的分子機制尚未完全闡明,因此,研究其中的分子機制對于尋找AS治療新藥具有重要意義。
MicroRNA(miRNA)是一種小的內源性非編碼RNA,其通過與靶mRNA的3'非翻譯區(UTR)結合,在轉錄后調控基因的表達〔2〕。miRNA在炎癥、細胞增生和脂質代謝等多種細胞機制調節中發揮關鍵作用,并參與AS的發生發展,如miR-217通過靶向SIRT1抑制巨噬細胞凋亡和炎癥反應〔3〕;miR-155通過誘導ERK1/2途徑激活和NLRP3炎性體活化,促進炎癥反應〔4〕。近期研究提示,miR-24在腫瘤〔5〕、心肌缺血再灌注損傷〔6〕和類風濕關節炎〔7〕等多種人類重大疾病中表達異常,表明miR-24與人類重大疾病的發生發展密切相關。為進一步探討miR-24對炎癥反應的調節作用及潛在的細胞機制,本研究通過建立體外Ox-LDL誘導人臍靜脈內皮細胞(HUVECs)炎癥損傷模型,探討miR-24對HUVECs炎癥損傷的影響。
1 材料與方法
1.1細胞株、試劑與儀器 細胞株:HUVEC購自武漢普諾賽生命科技有限公司。試劑:GV367慢病毒載體(上海吉凱基因公司);Qx-LDL(廣州奕元生物科技有限公司);細胞計數試劑盒(CCK-8,武漢博士德生物技術公司);Transwell小室(美國Corning);酶聯免疫吸附試驗(ELISA)試劑盒(日本同仁化學研究所);RNA提取、逆轉錄試劑盒和SYBR熒光定量PCR試劑盒(日本TAKERA公司);miRNA反轉錄試劑盒All-in-OneTM miRNA First-Strand cDNA Synthesis Kit(GeneCopoeia);兔抗ICAM-1、NF-κB、GAPDH和二抗(美國Cell Signaling Technology),PCR引物(生工生物有限公司),序列見表1。

表1 qRT-PCR引物序列
儀器:實時熒光定量PCR儀(原應用生物系統公司);蛋白電泳儀、轉膜儀 (美國Bio-Rad公司);Odyssey熒光掃描成像系統(美國LI-COR CXL)。酶聯免疫檢測儀Model-450(美國Thermo Forma公司)。
1.2miR-24慢病毒載體的構建與鑒定 委托上海吉凱基因化學技術有限公司以miR-24正義序列:5'-UGGCUCAGUUCAGCAGGAACAG-3',miR-24反義序列:5'-ACCGAGUCAAGUCGUCCUUGUC-3',陰性對照隨機序列:5'-GUCAUCAGUCGAGCUAGA-CGAG-3',設計miR-24高表達、anti-miR-24和miR-NC慢病毒載體。病毒載體上含嘌呤霉素抗性蛋白,可用嘌呤霉素篩選得到穩定細胞株。
1.3HUVECs的培養、慢病毒感染和實驗分組 用加有15%新生胎牛血清、1% 100 IU/ml青霉素和100 μg/ml鏈霉素的高糖培養液培養HUVECs。后續實驗均取對數生長期細胞。將HUVECs以細胞密度為5×104個/ml接種于6孔板,每孔2 ml,孵育過夜至細胞貼壁。上述3種慢病毒載體以感染指數(MOI)=70感染HUVECs。實驗分為空白組、Ox-LDL組、miR-24+Ox-LDL組、anti-miR-24+Ox-LDL組、miR-NC+Ox-LDL組。空白組正常培養,Ox-LDL組給予100 μg/ml Ox-LDL 處理24 h誘導細胞炎癥損傷,其余感染組細胞均先感染相應慢病毒載體繼而以同步法造模。應用不同濃度(0、25、50、100、200 μg/ml)Ox-LDL刺激HUVECs,并在12、24、48 h測定miR-24的表達量。
1.4CCK-8法檢測細胞增殖 將細胞以密度8×104個/ml接種到96孔板,每孔100 μl,待細胞貼壁后,更換無血清培養液繼續培養24 h,之后按設定實驗條件處理細胞,然后每孔加入培養液100 μl和 CCK-8試劑 10 μl,避光孵育1 h,在波長設定為450 nm的酶標儀上測定吸光度值(A450 nm),實驗重復3次。實驗組細胞活力=A450 nm實驗組-A450 nm調零組,空白組細胞活力=A450 nm空白組-A450 nm調零組。應用不同濃度(0、25、50、100、200 μg/ml)Ox-LDL刺激HUVECs,并于12、24、48 h測定A450 nm值。
1.5Transwell檢測細胞遷移能力 按設定實驗條件處理細胞,并將細胞以密度5×104個/ml接種于24孔板,培養箱中遷移24 h,0.1%結晶子染色10 min,磷酸鹽緩沖液(PBS)沖洗干凈,室溫晾干,然后在顯微鏡下觀察細胞遷移情況,并隨機拍照記錄,采用人工計數法進行細胞計數。
1.6ELISA檢測IL-6、TNF-α的表達 將細胞以密度1×105個/ml接種于24孔板,每孔500 μl,待細胞貼壁后,更換無血清培養液繼續培養24 h,之后按設定條件處理細胞,參照ELISA試劑盒說明書加入反應體系檢測IL-6和TNF-α蛋白濃度。
1.7實時熒光定量-聚合酶鏈反應(qRT-PCR)檢測IL-6、TNF-α、ICAM-1的mRNA與miR-24的表達量 將細胞以密度1×105個/ml接種于6孔板,每孔2 ml,待細胞貼壁后,更換無血清培養液繼續培養24 h,之后按設定條件處理細胞。參照RNA提取、逆轉錄試劑盒說明書提取總RNA,并逆轉錄為cDNA,以cDNA為模板,根據SYBR熒光定量PCR試劑盒說明書進行PCR擴增,ABI Prism 7300序列檢測PCR系統實時檢測。miR-24以U6為內參,IL-6、TNF-α與ICAM-1以GAPDH為內參,采用2-ΔΔCt法進行數據分析。
1.8Western印跡檢測ICAM-1與NF-κB信號通路蛋白表達 細胞處理方法同1.7。收集各組細胞,用RIPA裂解各組細胞,12 000 r/min、4℃離心20 min,取上清液,使用二喹啉甲酸(BCA)試劑盒進行蛋白定量。各組取40 μg蛋白進行十二烷基硫酸鈉-聚丙烯酰胺凝膠電泳(SDS-PAGE)并轉移至聚偏氟乙烯(PVDF)膜上,5%脫脂奶粉封閉1 h。加入P65、磷酸化(pP65)P65和ICAM-1相應Ⅰ抗稀釋液(稀釋比例1∶1 000),4℃孵育過夜。次日加入H+L熒光標記二抗(稀釋比例1∶10 000),室溫避光孵育1 h。利用雙色熒光掃描成像系統分析蛋白電泳條帶灰度值,ICAM-1以GAPDH為內參,蛋白相對表達量=目的蛋白灰度值/GAPDH灰度值;磷酸化P65以P65為內參,磷酸化蛋白相對表達量=磷酸化蛋白灰度值/總蛋白灰度值。
1.9統計學分析 采用SPSS25.0軟件進行ANOVA、LSD-t檢驗。
2 結 果
2.1慢病毒感染后miR-24的表達量 與空白組(1.00±0.00)相比,miR-NC組細胞內miR-24的相對表達量(1.07±0.09)差異無統計學意義(P>0.05)。與miR-NC組相比,miR-24組細胞內miR-24的相對表達量(1.85±0.15)顯著升高(P<0.05),anti-miR-24組細胞內miR-24相對表達量(0.44±0.10)顯著降低(P<0.05)。說明miR-24、anti-miR-24慢病毒成功感染HUVECs。
2.2Ox-LDL降低 HUVECs中miR-24的表達量 隨著Ox-LDL濃度的上升,miR-24的表達量逐漸降低;且隨著給藥時間的延長,miR-24的表達量也逐漸降低。在濃度為100 μg/ml作用24 h時,miR-24的表達量變化顯著。在此基礎上選用100 μg/ml Ox-LDL作用 HUVECs 24 h。見表2、圖1。

圖1 miR-24對HUVECs遷移的影響(×200)
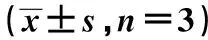
表2 不同濃度Ox-LDL對miR-24、HUVECs表達量的影響
2.3miR-24抑制IL-6、TNF-α和ICAM-1 mRNA的表達 與空白組相比,Ox-LDL組IL-6、TNF-α和ICAM-1 mRNA的相對表達量顯著上升(均P<0.05)。與miR-NC+Ox-LDL組相比,miR-24+Ox-LDL組IL-6、TNF-α和ICAM-1 mRNA的相對表達量顯著降低(均P<0.05),anti-miR-24+Ox-LDL組顯著升高(均P<0.05)。見表3。
2.4miR-24抑制IL-6、TNF-α、ICAM-1與NF-κB信號通路蛋白pP65的表達 與空白組比較,Ox-LDL組IL-6、TNF-α、ICAM-1與NF-κB信號通路蛋白pP65的表達量顯著上升(均P<0.05)。與miR-NC+Ox-LDL組相比,miR-24+Ox-LDL組IL-6、TNF-α、ICAM-1與NF-κB信號通路蛋白pP65的表達量顯著降低(均P<0.05),anti-miR-24+Ox-LDL組顯著升高(均P<0.05)。見表3,圖2。
表3 各組細胞中IL-6、TNF-α和ICAM-1 mRNA表達量、IL-6、TNF-α、ICAM-1及pP65的蛋白表達量比較

1~5:空白組,Ox-LDL組,miR-NC+Ox-LDL組,miR-24+Ox+LDL組,anti-miR-24+Ox-LDL組圖2 miR-24抑制HUVECs中pP65與ICAM-1蛋白表達水平
3 討 論
本研究miR-24對Ox-LDL誘導HUVECs炎癥損傷的影響及潛在分子機制顯示:miR-24抑制Ox-LDL誘導的內皮細胞炎癥損傷,抑制細胞增殖和遷移;miR-24通過抑制Ox-LDL誘導的NF-κB信號通路激活與IL-6、TNF-α、ICAM-1的分泌,參與炎癥反應進程。
VECs在免疫調節和炎癥反應中發揮重要作用。在受到炎癥刺激時,VECs被激活,分泌多種促炎細胞因子和細胞黏附分子,促進單核細胞、淋巴細胞及免疫細胞黏附并通過血管壁遷移至炎癥部位發揮炎癥效應。長期或過度炎癥反應引起的內皮功能障礙是AS發生發展的啟動事件之一。其中由Ox-LDL誘導的內皮炎癥反應與內皮功能障是促進動AS斑塊形成和發展的關鍵因素〔8〕。Han等〔9〕研究顯示,Ox-LDL可通過調控HDAC9的表達誘導內皮細胞凋亡,導致內皮損傷和炎癥發生,促進AS的發展。本研究結果提示Ox-LDL通過刺激NF-κB通路激活,誘導HUVECs損傷和炎癥反應發生。
miRNA是一種轉錄后基因調節因子,其差異性表達與AS的發生發展密切相關〔10〕。如miR-138在Ox-LDL誘導的HCAECs炎癥反應中下調,其通過抑制PI3K/Akt/eNOS信號通路激活,減輕HCAECs損傷和炎癥反應,阻滯AS的發展〔11〕。miR-24是一種富集于VECs并高度表達的miRNA。Di Gregoli等〔12〕研究發現,miR-24在AS斑塊中表達下調,其通過靶向抑制基質金屬蛋白酶-14的水解活性,抑制斑塊的形成。Zheng等〔13〕研究表明,miR-24在AS患者血清中表達水平顯著降低,其通過靶向抑制輸入蛋白-α3和NF-κB信號通路,抑制HUVECs炎癥反應。本研究結果顯示,經Ox-LDL刺激HUVECs炎癥損傷后,miR-24的表達水平顯著降低。因此推測miR-24的差異性表達與Ox-LDL誘導內皮細胞炎癥反應相關。
NF-κB是一種存儲于未活化細胞胞質中的快轉錄因子。正常生理情況下,NF-κB與其抑制性蛋白IκB結合處于非活化狀態;受到炎癥刺激時,IκB激酶復合物被激活,誘導NF-κB通路蛋白磷酸化,隨后NF-κB與IκB結合體解離并釋放NF-κB p65,NF-κB p65轉移至細胞核并與靶基因特異DNA識別位點結合,刺激細胞釋放IL-6、TNF-α、VCAM-1和ICAM-1等炎癥介質和細胞黏附分子,發揮促炎效應。過量的IL-6可誘導VEGF作用于VECs,導致血管生成活性和VECs通透性增加,引起炎癥細胞浸潤,脂質轉運和新內膜形成,促進脂質沉積,加重炎癥的發展〔14〕。TNF-α作為一種重要的炎癥介質,通過激活VECs,誘使VECs合成與分泌ICAM-1和IL-6等炎癥細胞因子,引發內皮功能障礙和血管損傷,導致血管完整性被破壞,參與炎癥反應過程〔15〕。ICAM-1是免疫球蛋白超家族中的一員,在VECs上存在組成性表達,通過與LFA-1(αLβ2)、Mac-1(αMβ2)整合素及各種肌動蛋白結合,誘使白細胞與內皮黏附,并促進白細胞穿過血管到達炎癥組織發揮促炎作用〔16〕。本研究結果提示,miR-24可能通過調控炎癥相關NF-κB信號通路的激活,及相關炎癥介質IL-6、TNF-α、ICAM-1的分泌,參與Ox-LDL誘導內皮細胞炎癥損傷的進程。
研究表明,miRNAs參與VECs的功能調節,并在AS的發生發展中發揮重要作用〔17〕。如miR-345-3p 在HUVECs中表達降低會激活TAK1/p38/NF-κB信號通路,從而導致內皮功能障礙和炎癥損傷,加重AS〔18〕;過表達miR-652-3p可通過抑制內皮修復基因Ccnd2的表達,抑制VECs增殖,加速AS病變〔19〕;miR-30-5p通過靶向TCF21抑制內皮細胞凋亡和炎癥反應,進而抑制AS的發展〔20〕。本課題組前期研究發現,miR-24促進氧化損傷下HUVECs的功能障礙,并抑制血管的形成〔21,22〕。本研究結果提示miR-24可能通過調節炎癥反應,抑制內皮細胞的增殖和遷移。
綜上,在Ox-LDL誘導HUVECs炎癥損傷模型中,miR-24可能通過抑制NF-κB信號通路的活化,阻滯內皮細胞分泌IL-6、TNF-α、ICAM-1,減輕HUVECs炎癥損傷,抑制細胞增殖和遷移。而下調miR-24的表達可加重HUVECs炎癥損傷,促進細胞增殖和遷移。
