國產注射用頭孢哌酮鈉舒巴坦鈉的質量評價
2022-02-19 08:46:34陳蓉馬冬陽蔡雪萍蘇靜華顧曉紅郭彬朱乃軍許丹郝剛邢以文
中國抗生素雜志 2022年1期
陳蓉 馬冬陽 蔡雪萍 蘇靜華 顧曉紅 郭彬 朱乃軍 許丹 郝剛 邢以文
(蘇州市藥品檢驗檢測研究中心,蘇州 215104)
頭孢哌酮(C25H27N9O8S2)為第三代廣譜半合成頭孢菌素,通過抑制胞壁黏肽合成酶,從而阻礙細胞壁黏肽合成,使細菌胞壁缺損,菌體膨脹裂解。能對抗多種β-內酰胺酶的降解作用,抗菌譜廣,對革蘭陽性菌及陰性菌均有作用[1]。舒巴坦鈉(C8H10NNaO5S)屬于半合成青霉烷砜類衍生物,為不可逆的競爭性β-內酰胺酶抑制劑,是繼克拉維酸后第二種用于臨床的β-內酰胺酶抑制劑,本身的抗菌活性弱,略強于克拉維酸,單用時僅對淋球菌和不動桿菌屬有殺菌作用,但其對多數β-內酰胺類抗生素耐藥菌株產生的β-內酰胺酶具有不可逆的抑制作用,因而可保護β-內酰胺類抗生素免受水解破壞[2]。兩種藥物合用,具有明顯的協同作用,其抗菌作用較單用頭孢哌酮高4倍[3]。
注射用頭孢哌酮鈉舒巴坦鈉(cefoperazone sodium and sulbactam sodium for injection)是頭孢哌酮鈉與舒巴坦鈉組成的復方制劑。最先由美國輝瑞公司研制,1986年在日本率先上市,1994年9月由大連輝瑞制藥有限公司獲批原研進口,1997年起國內生產企業相繼獲批上市。臨床上主要用于抗易感菌株引起的中度和重度感染治療,并限用于有明確藥敏試驗證據或重癥感染的患者。主要治療由敏感細菌所引起的呼吸道感染、泌尿道感染、腹內感染、敗血癥、腦膜炎、皮膚及軟組織感染、眼部感染、骨骼及關節感染等;預防因腹腔、婦科、心血管、骨科及整形手術所引起的手術后感染[4]。美國胸科學會/感染學會(ATS/IDSA)指南(2019年)推薦亞洲國家將注射用頭孢哌酮鈉舒巴坦鈉作為中重度院內獲得性肺炎(HAP)、呼吸機相關性肺炎(VAP)、多重耐藥菌株(MDR)、銅綠假單胞菌、肺炎克雷伯菌、不動桿菌屬等感染的初始經驗用抗生素之一。
注射用頭孢哌酮鈉舒巴坦鈉由頭孢哌酮鈉舒巴坦鈉原料直接無菌分裝制成,因此其原料的品質直接影響制劑的質量[5]。中國藥典2015年版二部[6]和日本藥典JP17版[7]收載了原料及其制劑,歐洲藥典(EP)9.0[8]僅收載了原料標準。該品種臨床使用非常廣泛,連續多年蟬聯國內醫療機構抗感染藥物使用量TOP1。不良反應發生數較高,常見的嚴重不良反應為皮疹、瘙癢、呼吸困難、胸悶、凝血障礙、過敏樣反應、寒戰、過敏反應、血小板減少[9]。隨著近年來“限抗令”的實施,對該品種的質量風險控制尤為重要。
目前國內共有注射用頭孢哌酮鈉舒巴坦鈉生產企業89家,批準文號352個,包括11種規格。國家藥品監督管理局將本品種列入2020年度國家藥品抽驗計劃。按照要求,我單位對國內39家企業生產的260批次注射用頭孢哌酮鈉舒巴坦鈉進行了法定檢驗和探索性研究,客觀評價該品種的質量現狀,查找可能存在的影響品種安全性和有效性的因素,提出質量標準完善建議,為企業提高生產工藝起到積極作用,同時也為上市后的藥品監管提供可靠的的技術支撐。
1 儀器與試藥
1.1 儀器
Waters Acquity H Class超高效液相色譜儀(美國Waters公司);Waters ACQUITY 2D UPLC液相色譜系統,Xevo G2-XS Q-TOF質譜系統配備MassLynx工作站(美國Waters公司);Agilent 7200 Accurate-Mass Q-TOF GC/MS配備NIST MS Search2.0工作站(美國Agilent公司);Thermo X-Series 2型ICP-MS,配備PlasmaLab工作站(美國Thermo Fisher Scientific公司);HACH TL2350濁度儀(美國Hach公司);Mastersizer 2000智能激光粒度儀(英國Malvern公司);XSE205DU型電子天平(瑞士Mettler Toledo公司);Direct-Q3型超純水機(美國Millipore公司);SK8210HP型超聲波清洗器(上海科導超聲儀器有限公司);XMTD-204型數顯式電熱恒溫水浴鍋(上海躍進醫療器械有限公司);Metrohm 915KF卡氏水分測定儀(瑞士萬通公司)。
1.2 試藥
對照品:頭孢哌酮、頭孢哌酮雜質A、頭孢哌酮雜質C(1-甲基-5-巰基四氮唑)、頭孢哌酮雜質F(頭孢哌酮S異構體)與舒巴坦、N-亞硝基二甲胺,以上均購自中國食品藥品檢定研究;頭孢哌酮雜質B、頭孢哌酮雜質D、舒巴坦雜質A、舒巴坦雜質C、舒巴坦雜質E、舒巴坦雜質F為EP對照品; 鉛、鎘、砷、汞、鈷、鎳、釩、硒、鋰、銻、鋇、鉻、鉬、銅、錫、鈀、鋅、鐵,以上均購自國家有色金屬及電子材料分析測試中心;BHT(2,6-二叔丁基-4-甲基苯酚)、八甲基環四硅氧烷均購自Sigma公司。
試劑:乙腈、四丁基氫氧化銨均為色譜純;冰醋酸、硝酸均為UP級;水為超純水,其余試劑均為分析純。
1.3 樣品
260批次注射用頭孢哌酮鈉舒巴坦鈉均為2020年國家藥品計劃抽驗樣品,涉及39個生產企業。企業覆蓋率43.8%、批準文號覆蓋率24.1%;抽樣覆蓋了全國22個省、4個直轄市、5個自治區,抽樣數量在各省之間分布均勻。其中從生產企業抽樣112批,經營企業抽樣139批,醫療機構抽樣9批。
2 實驗方法
2.1 法定標準檢驗
此次抽驗全部批次按照《中國藥典》2015年版二部注射用頭孢哌酮鈉舒巴坦鈉標準檢驗,部分項目涉及到國家食品藥品監督管理局注冊標準5個:YBH00672013、YBH11332006、YBH15742005、YBH28592005、WS1-(XG-002)-2005。
2.2 探索性研究
為全面評價注射用頭孢哌酮鈉舒巴坦鈉的質量,完善質控體系,探討工藝對產品質量的影響,結合企業調研結果、法檢結果、以及常見抗生素粉針劑研究方向等參考資料[10-12],圍繞藥品安全性和有效性兩個方面進行相關的探索性研究。
2.2.1 有關物質I
根據EP 9.0頭孢哌酮及舒巴坦項下有關物質方法,探索頭孢哌酮雜質A、B、C、D、F以及舒巴坦雜質A、C、E、F等雜質的分離,建立統一的UPLC方法進行檢查,結合雜質單標對照品定位和破壞試驗,考察樣品的雜質譜。
色譜條件:色譜柱:Waters Acquity UPLC HSS T3(2.1 mm×100 mm,1.8 μm);以0.05%三氟乙酸水溶液為流動相A,以乙腈為流動相B,按表1梯度洗脫;流速:0.4 mL/min;柱溫:35℃;檢測波長:230 nm;進樣量:2 μL。
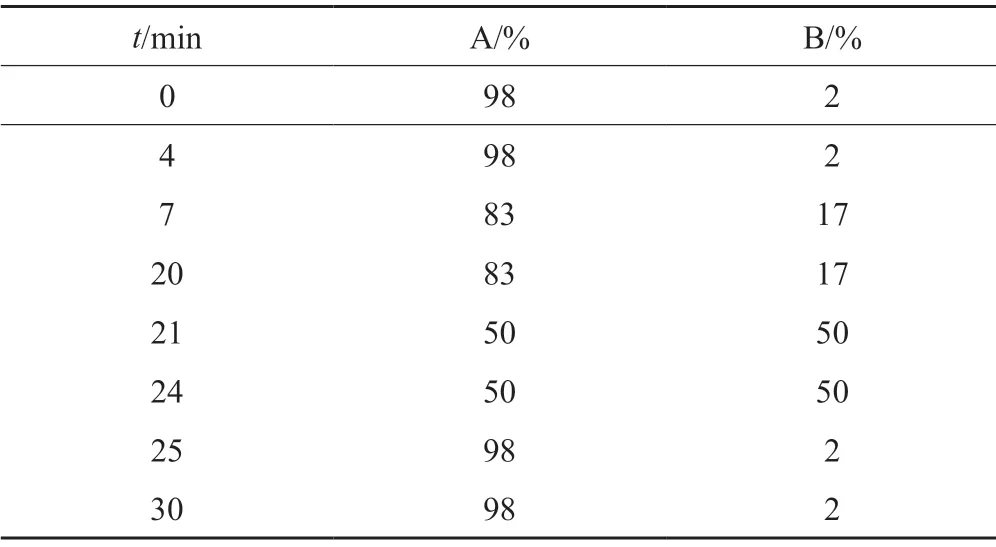
表1 梯度洗脫程序Tab.1 Gradient elution program
2.2.2 有關物質Ⅱ
引發過敏反應的過敏原是β-內酰胺類抗生素中的聚合物類雜質[13]。建立聚合物類雜質的定量分析方法,并采用二維柱切換超高效液相色譜-四級桿/飛行時間質譜(UPLC-Q/TOF-MS)聯用技術對其雜質進行結構推斷。
第一維色譜條件:色譜柱:TSK-gel G2000SWxl凝膠柱(7.8 mm×300 mm,5 μm);以磷酸鹽緩沖液(pH7.0)[0.005 mol/L磷酸氫二鈉溶液-0.005 mol/L磷酸二氫鈉溶液(61:39)]-乙腈(95:5)為流動相;流速:0.5 mL/min;柱溫:25℃;檢測波長:254 nm;進樣量:20 μL。第二維色譜條件:色譜柱:ACQUITY UPLC HSS T3(2.1 mm×100 mm,1.8μm);以0.005 mol/L甲酸銨水溶液為流動相A,以乙腈為流動相B,按表2梯度洗脫;流速:0.4 mL/min;柱溫:25℃;切換用定量環體積:250 μL。
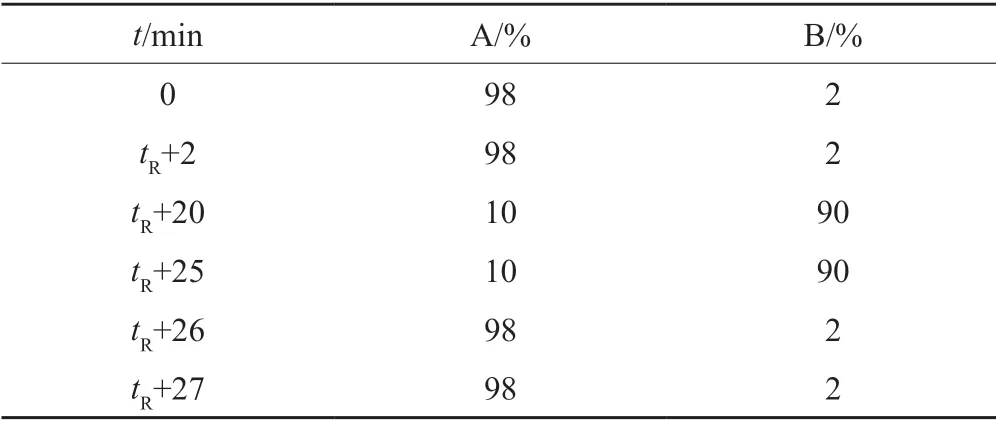
表2 梯度洗脫程序Tab.2 Gradient elution program
質譜條件:ESI離子源;正離子模式;一級質譜掃描范圍:m/z50~1500;離子源溫度:110℃;毛細管電壓:2.50 kV;錐孔電壓:40 V;去溶劑氣(N2)溫度:250℃;流量:600 L/h;錐孔氣流:50 L/h。
2.2.3 金屬元素的提取和遷移
玻璃由于本身的化學成分及生產工藝等因素,其中的金屬離子或陽離子團均有可能從玻璃中遷移出來,造成藥品中殘留無機金屬元素雜質。為了進一步研究樣品中金屬元素雜質的含量及與工藝等的內在聯系,開展了西林瓶及藥物中金屬元素的提取及遷移量研究。根據元素雜質可能的幾種來源,包括合成催化劑、玻璃包材常用元素及ICH相關要求,選擇了鉛、鎘、砷、汞、鈷、鎳、釩、硒、鋰、銻、鋇、鉻、鉬、銅、錫、鈀、鋅和鐵等18種元素作為考察對象,建立ICP-MS法考察39個廠家使用的西林瓶及108批制劑中上述金屬元素的提取及遷移量,評價西林瓶的安全性。
2.2.4 膠塞相容性
在藥物的貯存過程中,膠塞直接與粉末接觸,膠塞中的一些揮發性成分不可避免的與頭孢菌素發生相互作用,其與特定揮發性成分的吸附作用是導致注射劑澄清度變差的最主要原因。建立GC-MS法測定各廠家膠塞和制劑中的4種硅氧烷類和2,6-二叔丁基-4-甲基苯酚(BHT)等揮發性成分,采用濁度儀測定溶液的濁度值,同時選取兩家企業的頭孢哌酮鈉原料藥、舒巴坦鈉原料藥進行模擬實驗,與膠塞揮發物試劑(BHT和八甲基環四硅氧烷)一同密閉放置1、2和5d后,考察揮發性成分及濁度值變化,探討兩者之間的相關性,為膠塞的選擇提供參考。
2.2.5 粒度與粒度分布
藥物的粒子大小也是決定其溶解速率的因素之一,采用Malvern Mastersizer 2000激光粒度分析儀對39家企業260批制劑的粒度和粒度分布進行了測定。實驗參數:干法測定。背景及樣品的掃描時間為10 s;顆粒折射率1.52;顆粒吸收率0.1;分散壓力2.0 bar;進樣速率50%;遮光度0.5%~5%。
同時考查各廠家制劑復溶時間:取各廠家制劑1支,分別加入注射用水適量,使頭孢哌酮含量為125 mg/mL,置震蕩器上振搖溶解,轉速100 r/min,觀察并計時。
2.2.6 影響因素考察
參照中國藥典2015年版通則9001原料藥與制劑穩定性試驗指導原則,考察高溫(60℃,30%)、高濕(25℃,90%)、光照(25℃,4000Lx)3種條件下,39家生產企業的53批樣品放置1個月后的酸度、有關物質Ⅰ、有關物質Ⅱ等指標的變化情況。
2.2.7 含量均勻度
中國藥典2015年版四部通則0941含量均勻度檢查法項下規定,藥物間或藥物與輔料間采用混粉工藝制成的注射用無菌粉末均應檢查含量均勻度。雖然注射用頭孢哌酮鈉舒巴坦鈉制劑標準項下并未規定含量均勻度檢查,但雙組分不同配比的制劑,通過含量均勻度考察可以對藥品質量及分裝均勻性進行評價,使含量的分析結果更客觀。
色譜條件、系統適用性、對照品溶液配制等條件均同法檢含量測定。供試品溶液配制:取本品10瓶,分別溶解并定量稀釋制成約含頭孢哌酮0.5mg/mL溶液。對39個廠家260批不同規格注射用頭孢哌酮鈉舒巴坦鈉進行含量均勻度檢查,并對結果進行分析。
3 結果與分析
3.1 法定檢驗結果
按現行質量標準檢驗,39家企業的260批次樣品均符合規定,合格率100%。本次抽樣樣品生產時間介于2018年11月至2020年6月之間,由圖1可知,檢驗結果表明該產品質量與生產時間具有一定的相關性,隨著樣品從生產距離檢驗時間延長,頭孢哌酮雜質A、頭孢哌酮雜質C、有關物質Ⅱ雜質呈變大的趨勢,溶液的澄清度從淺于0.5號濁度標準液逐漸加深至淺于1號濁度標準液,溶液的顏色由淺于黃色或黃綠色2號標準比色液逐漸加深至淺于黃色或黃綠色4號標準比色液。本次從不同抽樣點抽取同企業同批號樣品12組(24批),對其酸度、水分、頭孢哌酮雜質A、雜質C、其他最大單雜、其他總雜、頭孢哌酮含量、舒巴坦含量8個檢驗指標結果進行了SPSS配對t檢驗,結果表明同批號樣品檢驗結果間無顯著差異(P>0.05),表明在規定的儲藏條件下(密閉,在涼暗干燥處)保存,樣品質量與抽樣地無明顯相關性,具體結果見表3。
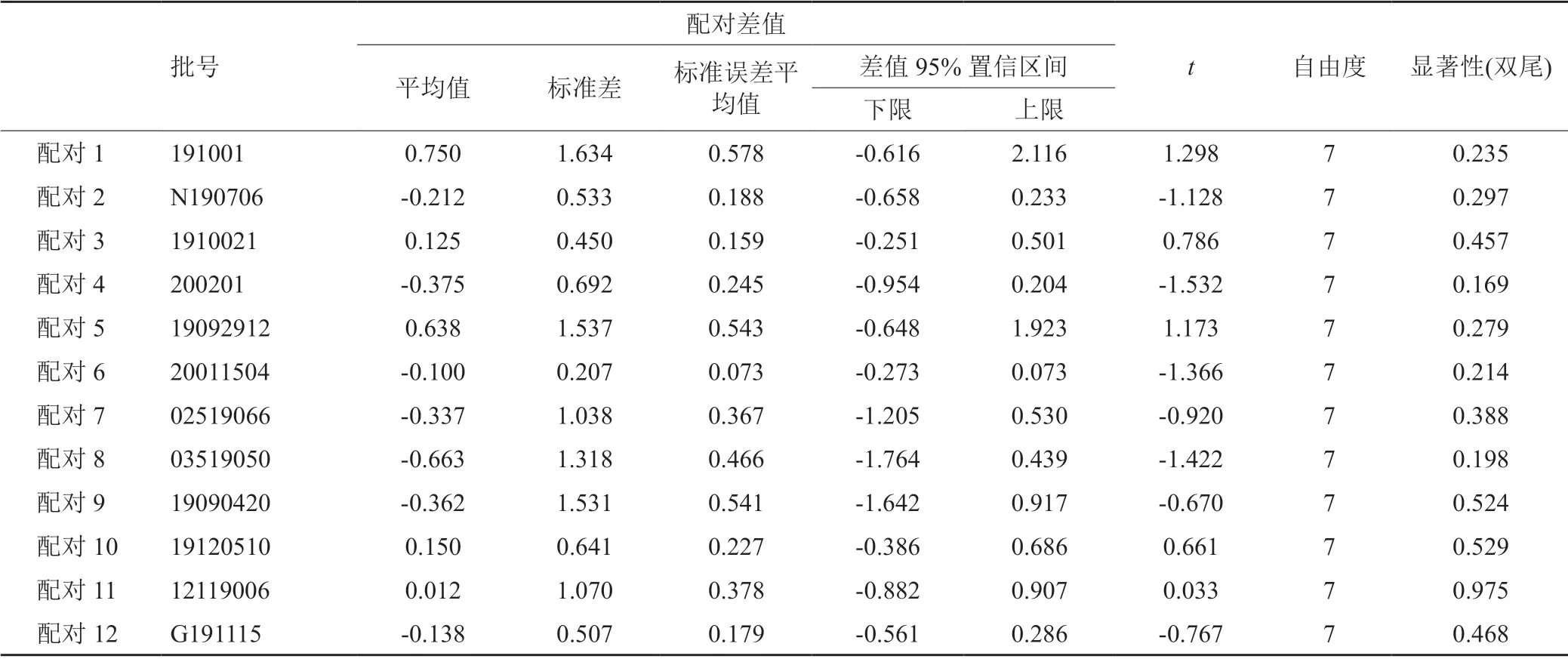
表3 同批號配對樣本檢驗Tab.3 Paired sample test of same batch samples
3.2 探索性研究結果
3.2.1 有關物質I
《中國藥典》2015年版二部僅控制頭孢哌酮雜質A、頭孢哌酮雜質C兩個已知單雜及其他單雜、其他總雜,典型供試品色譜圖如圖2。采用“2.2.1”項下建立的雜質譜分析方法共檢出9個雜質峰,各雜質峰與2個主峰(峰3為舒巴坦峰,峰9為頭孢哌酮峰)分離度較好,如圖3~4,溶劑空白表明基線基本無干擾,9種成分在各自標曲范圍內均呈現良好線性(r≥0.9998),精密度、重復性、回收率均滿足要求(RSD<2.0%),頭孢哌酮雜質A、B、C、D、F和舒巴坦雜質A、C、E、F檢出限分別為:0.022、0.313、0.093、0.316、0.319、0.029、0.836、2.194和0.265 μg/mL。經檢測260批樣品:有27批檢出了舒巴坦雜質A(峰1),按舒巴坦標示量計為0.07%~0.18%;有108批檢出了頭孢哌酮雜質F(峰10),按頭孢哌酮標示量計為0.05%~0.22%;頭孢哌酮雜質A(峰7),按頭孢哌酮標示量計為0.3%~1.0%。頭孢哌酮雜質C(峰2),按頭孢哌酮標示量計為0.1%~0.5%;其他最大單雜按自身對照法計為0.1%~0.5%;其他雜質總和按自身對照法計為0.4%~2.4%。
由圖5可知,與現行標準有關物質檢測方法相比,新建方法的頭孢哌酮雜質A和頭孢哌酮雜質C結果差異不明顯;其他最大單雜、其他雜質總和的檢出數量和含量均明顯增加,且檢出的雜質含量平均偏差小于法檢方法。由于雜質多樣性以及檢測手段的局限性,UPLC法仍需要進一步進行研究論證,方可作為該品種有關物質檢查標準提高的補充。
3.2.2 有關物質Ⅱ
按“2.2.2”項下方法檢驗,測定結果在0.81%~2.11%之間。由圖6可知,在TSK柱的測定條件下,共檢出4種雜質,經LC-MS分析,雜質1和雜質2未檢出明顯的雜質峰,未做定性推測;雜質3檢出1個雜質峰,雜質4共檢出4個雜質峰,各雜質可能的化學結構及來源分析:雜質3可能為頭孢哌酮與其降解雜質及工藝雜質形成的三聚物,雜質4a可能為頭孢哌酮降解物或衍生物與頭孢哌酮雜質E的聚合物,雜質4b、4c、4d為同分異構體,可能為頭孢哌酮與其降解雜質及工藝雜質二聚物,具體雜質推測見表4。
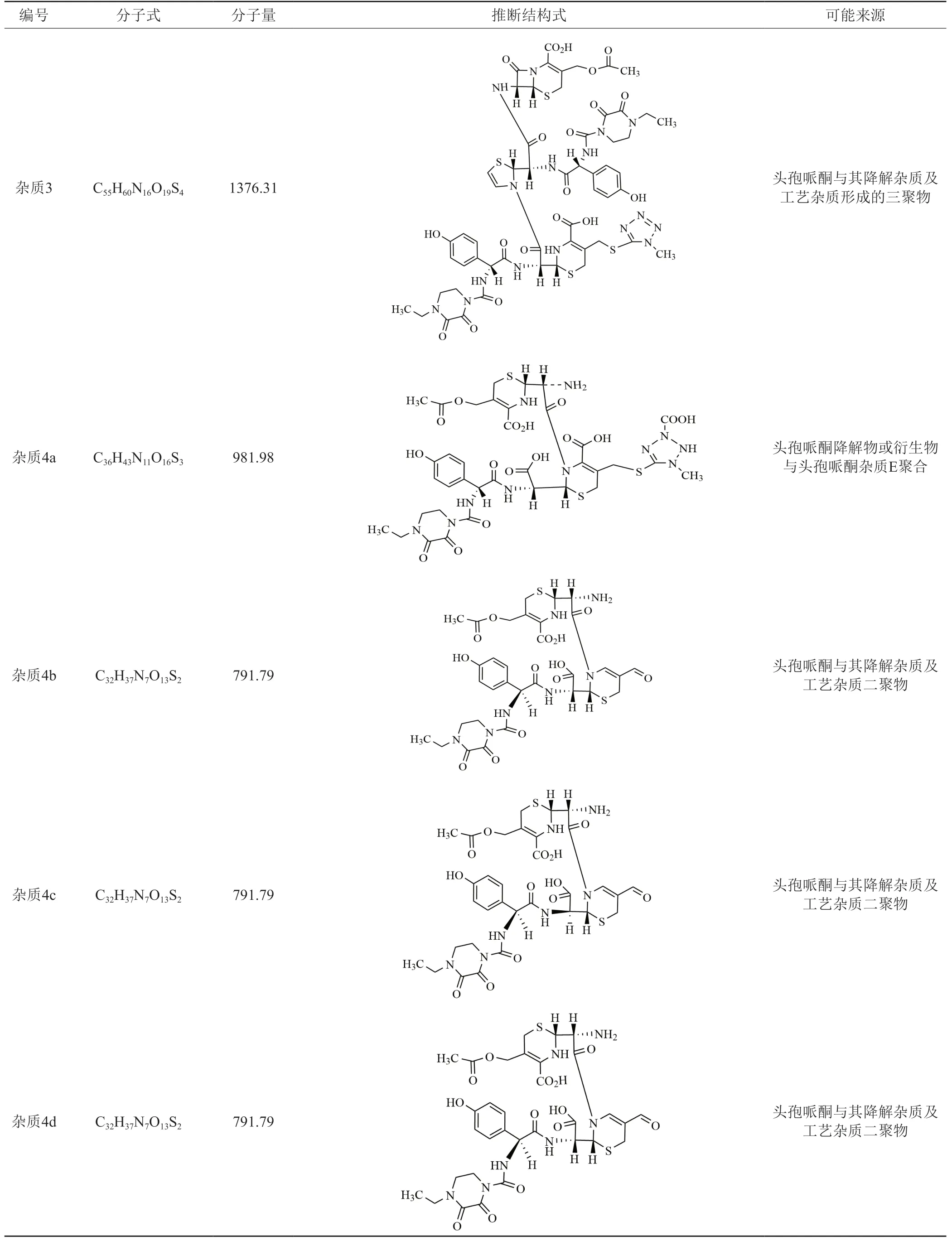
表4 各雜質可能的化學結構及來源分析Fig.4 Possible chemical structures and sources of impurities
現行標準僅YBH00672013對其聚合物類雜質(有關物質II)進行了控制。由上述方法測定發現,主峰前的色譜峰并不都是聚合物雜質,個別色譜峰也不是純峰,而是多個化合物(雜質)的混合物[14]。有關物質Ⅰ不能全面反映該品種的全部雜質情況。有關物質Ⅱ既能檢查聚合物類雜質,又可作為有關物質Ⅰ檢查的一個補充,可完善雜質譜,以減少不良反應的發生。
3.2.3 金屬元素的提取和遷移
39個廠家西林瓶離子檢測結果可以得出,除鈷、鉬、汞、鈀、硒5種離子均無檢出,其余13種離子部分廠家有檢出。其中低硼硅玻璃管制注射劑瓶的Ba、Zn、Fe含量顯著高于鈉鈣玻璃模制注射劑瓶。鈉鈣玻璃模制注射劑瓶浸出的金屬離子數量和含量均較低硼硅玻璃管制注射劑瓶偏低,理論上來說相對更安全。與《化學藥品注射劑與藥用玻璃包裝容器相容性研究技術指導原則(試行)》及ICH Q3D “元素雜質指導原則”規定的PDE(每日允許暴露量)相比,39家企業采用的西林瓶浸出離子含量均符合規定。108批制劑離子檢測結果顯示,除銻、鈀、硒3種離子均無檢出,其余15種離子大部分廠家均有檢出,但離子濃度均無明顯的變化趨勢,說明藥液與注射劑瓶內表面之間未發生明顯遷移。可認為浸出物的量不會改變藥品的安全性,對患者的安全性風險小。表明目前采用的各類西林瓶內包材與該品種制劑相容性較好。
3.2.4 膠塞相容性
由圖7可知,濁度較大的樣品,其揮發物中含有的硅氧烷類和BHT越多,表明制劑溶液的濁度值與硅氧烷類和BHT具有正相關性,提示生產企業在選擇膠塞時,應著重考察膠塞中硅氧烷類和BHT的含量。模擬吸附試驗表明,頭孢哌酮鈉吸附的BHT含量與濁度值有一定的相關性,舒巴坦鈉吸附的硅氧烷類和BHT含量與濁度值幾乎沒有相關性,即硅氧烷類和BHT對溶液澄清度的影響主要作用于頭孢哌酮鈉。結合法檢和探索性研究得出,影響溶液澄清度的因素主要為藥品與膠塞作用的時間、規格、膠塞種類等。頭孢哌酮鈉與膠塞作用時間延長,濁度值增大;小規格制劑與膠塞接觸更為充分,隨著時間的推移,膠塞中的硅氧烷類和BHT遷移到藥物粉末中的更多,濁度值越大;在膠塞選擇時盡可能選擇覆膜膠塞。國內兩家企業的0.5g規格制劑,采用濁度儀法測定溶液的澄清度,均超過了1號濁度標準液,由于同一廠家不同規格制劑均使用相同膠塞,藥物粉末與膠塞接觸而發生的相互影響是一樣的,但在澄清度時加水量不一樣,規格小的制劑可能澄清度較其他規格的差,存在影響藥品安全性的質量風險,應密切注意該風險。
3.2.5 粒度與粒度分布
由圖8復溶時間研究表明,原研樣品(9號)在60秒內全部溶解,大部分企業產品在60~120 s內溶解,小部分廠家制劑120s后仍有殘留未溶解。目前該品種國內主要有3大原料藥生產廠家,分別為:SZDR、QLAT、ZHLB,通過比對各廠家原料藥來源發現,編號為1、3、4、6、10、12、17、15、18、19、25、27、28、29、30、31、33的制劑原料藥來自于SZDR,普遍溶解較快;編號為2、8、22、23、26、34的制劑原料藥來自于ZHLB,溶解時間最長;其余來自QLAT和其他原料藥廠家,溶解時間多在1~2 min內。推測不同廠家制劑復溶行為存在差異,可能與其原料藥不同有關。
各廠家制劑的粒度分布范圍為D(0.1)=0.8~4.3 μm,D(0.5)=8.7~63.6 μm,D(0.9)=122.1~581.6 μm,原研藥品的90%粒徑范圍為197.8~361.9 μm,最大粒徑373.6 μm,來自SZDR原料藥的制劑企業的90%粒徑范圍為122.1~336.4 μm,最大粒徑380.4 μm,來自QLAT原料藥的制劑企業的90%粒徑范圍為169.4~412.4 μm,最大粒徑441.7 μm,來自ZHLB原料藥的制劑企業的90%粒徑范圍為236.4~581.6 μm,最大粒徑612.6 μm。原研藥品的粒徑比國產藥品的更小且分布較為均勻集中,推測可能更利于藥品的溶解,結果見表5。D(0.5)和D(0.9)的分布與頭孢哌酮和舒巴坦的配比具有相關性。如圖9所示,同一廠家,頭孢哌酮與舒巴坦配比為2:1規格的粒徑通常大于1:1規格。粒度分布考察可為處方配比及制劑工藝優選提供依據,提示企業在生產過程中進一步加強對原料和生產工藝的控制。
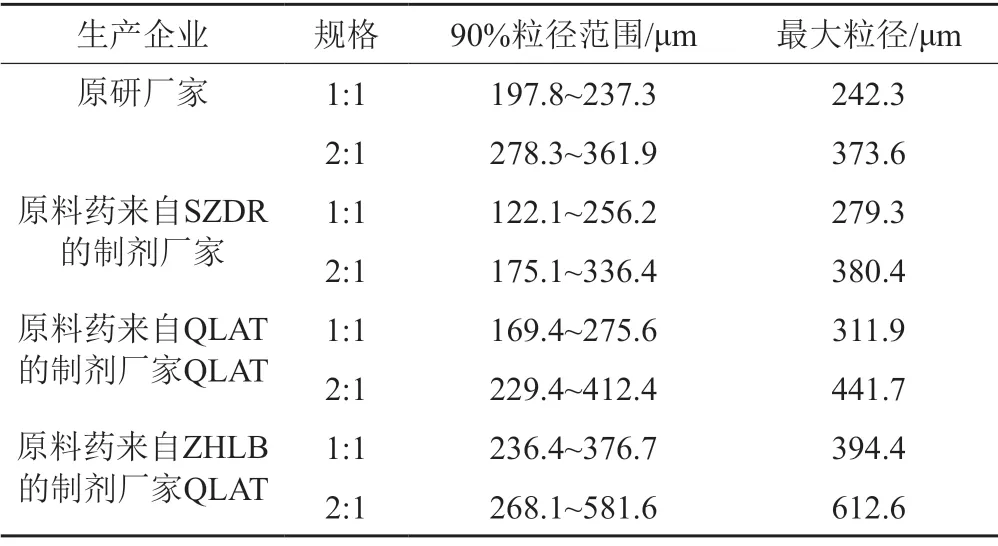
表5 粒徑分布分析結果Tab.5 Analysis of the particle size distribution of the samples
3.2.6 影響因素考察
高溫、高濕、光照處理后的樣品pH值均下降,其中高溫影響最顯著,高濕和光照結果相當;頭孢哌酮雜質A、頭孢哌酮雜質C、其他最大單雜、其他總雜、頭孢哌酮雜質F、舒巴坦雜質A 6個指標受高溫影響顯著增加;除舒巴坦雜質A對高濕和光照敏感外,其余5個指標在經過高濕或光照處理后,變化趨勢不明顯;高溫處理后的樣品有關物質Ⅱ含量顯著增長,高濕處理后基本沒變化,光照處理后略有增加。綜上所述,溫度是影響pH、雜質的最關鍵因素,提示生產、經營、使用各環節在儲存過程中必須注意溫度監控。
3.2.7 含量均勻度
259批制劑兩種主成分的含量均勻度結果良好,A+2.2S均小于15.0;一廠家1批規格2.25g(2:1)制劑的舒巴坦含量均勻度A+2.2S超標,可能是混粉分裝工藝存在問題,同時發現該廠家藥品的粒徑分布也相對較大,頭孢哌酮與舒巴坦顆粒大小的懸殊也容易導致混粉不均勻。如圖10,頭孢哌酮和舒巴坦含量總體呈正態分布,其中頭孢哌酮分布的中心點偏向于100%的左側,而舒巴坦分布的中心點則偏向于100%的右側。推測可能是生產時頭孢哌酮投料量可能未達到100%,而舒巴坦則可能超過100%;抑或是頭孢哌酮更容易降解,導致測得的含量低于100%。含量均勻度考察能更全面客觀地反映產品含量的真實情況,尤其適用于雙組分混合比例差距較大的粉針劑品種,因此建議在注射用頭孢哌酮鈉舒巴坦鈉質量標準中增加含量均勻度檢查。
4 結論
目前,注射用頭孢哌酮鈉舒巴坦鈉的法定質量標準基本合理,限度較為合適,能夠較好的控制本品的質量。按現行標準檢驗,本次抽驗的39家生產企業260批樣品,均符合標準規定。探索性研究修訂了有關物質檢查、增加了含量均勻度和有關物質Ⅱ等項目。采用擬修訂的標準對260批樣品檢驗,合格率99.6%。產品總體質量較好。
注射用頭孢哌酮鈉舒巴坦鈉為我國抗感染類藥物臨床使用量排名第一的品種,不良反應也相對多發。本品生產廠家繁多,但制劑工藝相對簡單統一,此次抽驗涉及中國藥典2015年版二部及5個企業注冊標準,上述標準主要區別在于部分企業在執行中國藥典2015年版二部的基礎上,按各自企業注冊標準執行異常毒性項目,且部分項目執行的限度不一致。各標準之間無較大差異,不利于整體質量控制,均未能有效地控制有關物質和聚合物雜質,缺乏含量均勻度等檢查。探索性研發現,產品在生產工藝、內包材、說明書等方面存在缺陷,臨床用藥有安全隱患。因此建議企業有條件時:①對法定標準進行規范統一;②修訂有關物質檢查方法和控制限度;③增訂包含聚合物的有關物質Ⅱ檢查項;④對其產品的分裝工藝進行研究,提高批間穩定性;⑤針對小規格制劑膠塞進一步開展包材相容性研究;⑥進行原料藥晶型和基因毒性雜質研究;⑦盡快開展一致性評價工作。
此外,該品種目前共有批準文號352個,本次抽樣僅涉及不足三成的文號,未抽到的大多數批準文號可能已經閑置,造成的不僅是社會資源的浪費,而且企業長期不生產,偶爾生產可能會存在質量風險,對藥品生產監管、上市藥品再評價、不良反應監測非常不利。因此建議企業梳理批準文號,對于長期不生產的品種應當查找原因;建議藥監部門對此類情況進行排查,加強監管,保障藥品質量。
