甲烷催化燃燒反應機理及催化劑研究進展
2022-02-21 06:28:14楚培齊王賽飛趙世廣鄧積光劉雨溪段二紅
燃料化學學報 2022年2期
楚培齊 ,王賽飛,2,* ,趙世廣 ,張 依 ,鄧積光 ,劉雨溪 ,郭 萌 ,段二紅,2,*
(1. 河北科技大學 環境科學與工程學院,河北 石家莊 050018;2. 揮發性有機物與惡臭污染防治技術國家地方聯合工程研究中心,河北 石家莊 050018;3. 北京工業大學 環境與能源工程學院,北京 100124)
能源供應是目前世界經濟增長和社會發展的主要關切點之一,雖近些年新能源發展勢頭強勁,但是不可否認的是化石資源(例如煤炭和天然氣)仍是電力、冶金和化工等國民產業能源“頂梁柱”[1]。然而,隨著煤炭開采量和天然氣使用量的增加,相應的問題也隨即出現。中國作為世界上最大的煤炭生產國和消費國,在煤炭開采過程中常伴隨著大量瓦斯的釋放,其中,70%是以風排瓦斯(指CH4濃度不超過1%的混合氣)的形式直接排放[2,3]。與此同時,天然氣因其具有較高熱值和豐富的儲量,在主要能源消費中占據了最大的增量并廣泛應用于工業生產及移動源領域。但是,作為天然氣的主要成分,未燃燒充分的CH4直接排放不僅造成資源浪費還加劇了大氣環境的污染。CH4是一種可以引發強烈溫室效應的氣體,根據全球變暖潛能值顯示,其增溫潛能是CO2的40倍,相應產生的溫室效應是等量CO2的21?26倍,對溫室效應的貢獻巨大[4?6]。特別是在當今減排節能的大環境下,全球氣候變化和溫室效應已經成為環境領域關注的重點,2021年中國“兩會”的政府工作報告中,將“扎實做好碳達峰、碳中和各項工作”列為重點工作之一,因此,CH4高效治理和利用成為大氣環境領域的重要研究內容。
根據實際工況和環境條件,甲烷的治理方法可分為多種,其中,催化燃燒法能在較低溫度下將CH4完全氧化為CO2和H2O,并且同時充分利用CH4能源。相較于吸附法、吸收法等技術手段,該方法具備效率高、壽命長、無二次污染等優勢[7],被認為是處理CH4的有效方法之一,成為當下研究的熱點[8]。Wang等[9]和張洪雁等[10]綜述了各類催化劑在甲烷催化燃燒應用以及多種因素對催化劑的影響,但是還未見針對各類催化劑有效提升活性、穩定性和抗毒性方法的綜述報道。因此,本文總結了近年來甲烷催化燃燒機理研究進展和催化劑性能改進方法及應用。從典型的貴金屬材料的熱穩定性和抗中毒性的提升,到金屬氧化物的活性增強分別作以概述,最后對未來催化劑材料的發展做以展望。
1 甲烷催化燃燒過程
CH4催化燃燒是一種強烈且不可逆的氧化反應,其反應方程式如下:
根據反應式可知,CH4氧化反應是一種強放熱反應,CH4分子中的碳在高溫下會被氧化為CO2,但是與此同時還伴隨著副反應的發生,例如甲烷-二氧化碳重整、CH4部分氧化和水氣變換反應,如式(2)、(3)、(4)所示:
根據反應方程式可知,CH4氧化在熱力學上是有優勢的,但是由于CH4分子具有四個完全相同且對稱的C–H鍵,每個C–H鍵擁有極高的鍵能(439 kJ/mol),這就使得CH4分子不易被活化,需要在較高的溫度下才能使其被氧化[11]。CH4催化氧化反應速率隨溫度而變化,如圖1所示[12],CH4催化燃燒反應經歷四個過程:A區域為表面反應動力學區(≤ 300 ℃);B區域為點火區;C區域為傳質控制區,其反應速率受表面反應和傳質過程共同影響(B、C兩區域統稱中溫區(300–800 ℃));D區域為高溫均相反應區(800–1500 ℃)[13]。CH4作為高穩定烷烴分子,若想獲得較高反應速率且避免高溫產生的二次污染,應將反應溫度控制在B區。同時,實際工況條件下的氣體還包含硫、氯、水汽等成分,易與催化劑反應位點結合而導致失活。因此,甲烷催化劑應具有低溫高活性、高熱穩定性和一定的抗水抗中毒能力。
2 甲烷催化燃燒機理
CH4分子在溫和條件下難以激活,為了實現其中低溫完全轉化,甲烷催化燃燒機理和動力學的研究對于指導催化劑的設計和反應工藝優化是必要的。關于CH4催化氧化的反應機理,按照不同氧物種的參與形式,常規認為存在四種機理,表面吸附氧主導的Langmuir-Hinshelwood(L-H)機理和Rideal-Elecy(R-E)機理,晶格氧主導的Mars-Van Krevelen(MVK)機理[14],及兩者共同主導的Twoterm(TT)機理。
L-H機理:認為分子氧在催化劑表面吸附活化能明顯小于CH4分子,分子氧優先吸附在催化劑表面形成吸附氧。同時,吸附氧比分子氧更容易和CH4分子結合,使其中的氫分子脫離,CH4穩定結構破壞,生成活潑的甲基自由基,從而促進CH4氧化。L-H機理主要用于貴金屬及其負載的氧化物體系[15]。
R-E機理:CH4分子首先和催化劑中的晶格氧結合,導致CH4分子中的C–H鍵斷裂形成自由基后被氧化。同時,CH4分子和催化劑結合過程中消耗晶格氧,產生呈弱電性的氧空位,吸附分子氧以補充催化劑中晶格氧的損耗[16]。
MVK機理:表面氧反應和晶格氧遷移,反應路徑可以分為以下三步[17?19]:
第一步,氣態CH4分子在催化劑活性位點上發生表面吸附,形成吸附態CH4。
第二步,吸附態CH4分子被表面晶格氧破壞,進而形成 CH+3,最后被氧化為吸附態CO2、H2O,同時形成氧空位,該步驟可描述為催化劑還原。
第三步,吸附態CO2、H2O從表面脫附形成氣態CO2、H2O,同時內部晶格氧遷移到表面,氧空位被表面吸附氧重新填充,該步驟可描述為催化劑再氧化。
相較于L-H機理,R-E機理和MVK機理主要適用于非貴金屬氧化物催化劑體系。
對于CH4催化燃燒反應,第一個C–H鍵的活化與斷裂為控速步驟,當首個C–H鍵斷裂,產生CH3基團和H自由基,引發鏈式反應[20,21]。針對上述反應機理,決定步驟的影響因素有所不同。對于L-H機理和R-E機理,其速率決定步驟一般被認為是表面反應,反應速率與催化劑表面吸附氧和過渡離子的電子性質有關。相反的是,MVK機理被認為是界面反應,其速率決定步驟與晶格氧空位相關。隨著甲烷催化燃燒機理研究的不斷深入,單一的機制描述催化劑上的反應過程存在一定局限性。Two-term(TT)機理提出一種“混合機制”—隨溫度的變化,表面吸附氧和結構中晶格氧兩種氧化途徑同時參與反應,對CH4的催化燃燒機理解釋較為完整,目前得到了廣泛地認可[11]。
Two-term機理主要過程如下:第一,CH4與催化劑表面氧相互作用形成氧空位;第二,催化劑中的氧空位促進氣態氧的吸附和解吸,同時促進晶格氧從結構內向表面擴散;但由于O2分子的活化比CH4中C–H鍵的活化更為容易,所以應先發生氧交換反應。這兩種途徑都能提高催化活性。Wang等[22]選取鈣鈦礦催化劑,并利用La基鈣鈦礦的Op帶中心和B位金屬陽離子d帶中心的相對費米能級(EF)作為活性描述符來解釋“Twoterm”機理。如圖2所示,當LaFexCo1?xO3的EF為負時,催化劑金屬性較強,鈣鈦礦為表面催化模型,表面氧起作用,低溫催化活性較好;當EF為正時,鈣鈦礦為界面催化模型,晶格氧起作用,高溫催化活性較好。值得注意的是,該項研究基于在以往對于尖晶石的研究之上,發現尖晶石催化一般遵循某一種反應路徑(不管是面上的R-E模型還是面內的MVK模型),而且如圖2(b)所示,在某一反應路徑占主導的溫度范圍內,遵循主導反應路徑的催化劑效率更高;相反,那些遵循另一種機制的催化劑效率弱。即催化劑本征活性與反應溫度有關[23]。這說明Two-term機理不僅限于鈣鈦礦型催化劑,尖晶石類催化行為也同樣適用,逐漸成為主流和完整的催化機理。
3 甲烷催化燃燒催化劑
CH4催化氧化對于催化材料性質的要求較高。一方面結構穩定,難于活化;另一方面氧化反應中較高的熱量釋放會導致催化劑的燒結。除此之外,考慮到催化劑在工業上的實際應用,CH4氧化物為CO2和H2O,且實際工況中包含水蒸氣和SO2,為此催化材料需要具備以下特征:低溫高活性,有效減少氮氧化物的產生,同時降低裝置要求;高熱穩定性,在較高溫度保持催化活性;抗毒抗水性;價格低廉等[24,25]。CH4催化氧化材料方面的報道有很多,主要有貴金屬催化劑、非貴金屬氧化物。表1給出了各類催化劑用于甲烷催化燃燒的實驗條件和催化性能,以t90(甲烷轉化率達到90%時對應溫度)作為評價標準。
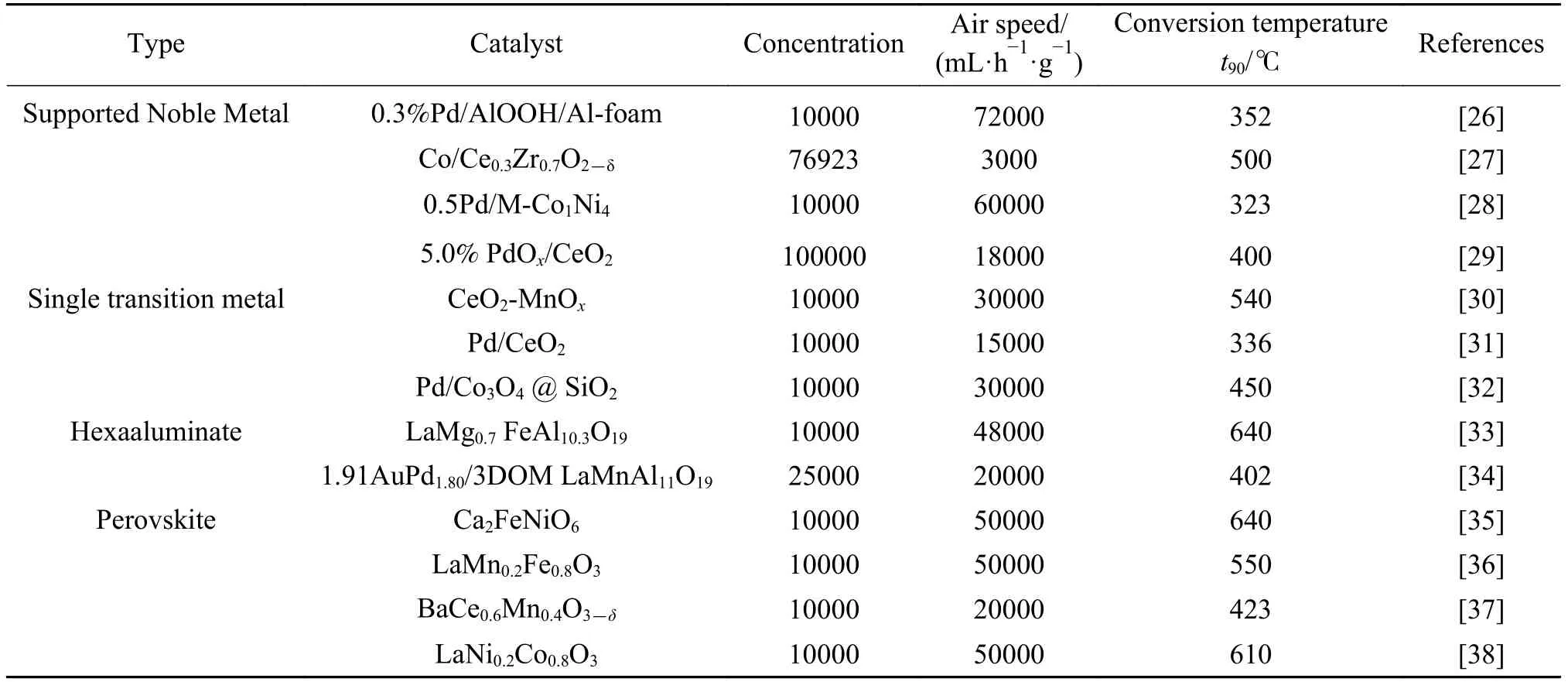
表1 催化劑催化燃燒CH4性能Table1 Comparison of CH4 catalytic combustion performance of catalysts
3.1 負載型貴金屬催化劑
貴金屬催化劑由于具有低溫高催化活性等優勢而得到廣泛研究。但是,考慮到貴金屬催化劑本身熱穩定性等問題,需要對貴金屬催化劑進行負載預處理。目前,用于CH4催化燃燒的貴金屬催化劑主要以Pd、Pt、Rh、Au為活性組分[39,40],金屬氧化物作載體。由于金屬氧化物具備比表面積大的優勢,有利于貴金屬活性組分在表面上呈現高分散狀態,暴露盡可能多的活性位點從而提高催化活性,此外貴金屬和載體之間的協同作用也可提高整體熱穩定性[41,42]。Pd基催化劑被認為是對CH4催化燃燒具有最高催化活性的貴金屬,因此成為主要研究對象[43,44],而對于金屬氧化物載體,一般多用CoOx、NiO、ZrO2和CeO2等。下面對研究較為深入的Pd、Pt、Au基貴金屬催化劑進行總結。
3.1.1 Pd基催化劑
Pd基催化劑已經得到了深入研究和廣泛應用,但對其在CH4氧化過程中的活躍狀態(Pd、PdO、Pd0/PdOx)還一直存在爭論,目前,PdO作為活性相得到了大多數專家學者的認可。Matam等[45]針對Pd基催化劑的活性位點進行了研究,通過XANES原位監測Pd/Al2O3在甲烷燃燒過程中Pd行為變化發現活性物種的性質取決于反應溫度。在677 ℃以下時,PdO是主要的催化活性位,并且催化性能取決于PdO粒徑;而當到677 ℃以上時候,還原后的Pd也能起到催化作用。這就表明Pd氧化態和還原態都可催化甲烷氧化,只是在不同溫度下機理和效果不一致。
近些年來,對于Pd基催化劑活性提升主要集中在提高載體比表面積或添加稀土金屬和Pd相互作用來增強活性位點的分散性和穩定性。Al2O3的物化性質穩定性和高比表面積成為了載體的普遍選擇,但是在高溫條件下高比表面積這一優勢極易被破壞,所以近些年各種載體材料被開發應用。Ding等[46]報道了合成具有高比表面積的Kappa相和四方相的CeZrO4固溶體,然后負載Pd催化劑用于CH4催化燃燒,發現Pd/k-CeZrO4中的PdOx粒徑更小,活性物種PdO含量更高,其氧遷移速率遠高于Pd/t-CeZrO4,在催化燃燒中具有優越的催化活性。稀土金屬添加也可有效穩定活性組分,例如La、Zr、Ce、Pr等,Cargnello等[47]率先提出一種新思路,如圖3(a)所示,即先形成Pd@CeO2核殼結構保護貴金屬組分后負載Al2O3上,當催化劑溫度到達850 ℃以上時,Pd相仍然穩定存在,沒有觀察到Pd-PdO轉化導致的失活現象,并且增強的金屬-載體相互作用導致甲烷氧化效果突出,說明Ce可以阻止PdO還原并促進Pd再氧化。
3.1.2 Pt基催化劑
雖然Pd基催化劑被認為是催化燃燒的最佳選擇之一,但是燒結和SO2中毒一直是待解決的問題。Pt基相對于Pd基催化劑,更適合高級烷烴的催化選擇,具備更高的耐毒性,甚至在一定環境下SO2會促進甲烷氧化。Kylhammar等[48]觀察到Pt/CeO2在SO2存在下,300–450 ℃時CH4氧化得到增強,但會隨著溫度升高逐漸減弱到最后轉為抑制氧化過程。通過原位紅外表征等表明,在CeO2載體上硫酸鹽形成過程中伴隨著氧空位產生防止氧溢出到Pt表面,從而促進甲烷轉化率瞬時增加。Chen等[49]充分結合Pd低溫高催化性能和Pt對SO2耐毒性,設計了Pd-Pt/MgAl2O4催化氧化CH4,Pt可以有效穩定活性物種PdO中的Pd,同時抑制PdO分解為反應性低的Pd0,同時雙貴金屬之間的電子相互作用增強了催化劑整體的水熱穩定性。由此可見,無論是以何種方法來提升貴金屬催化劑催化活性,都是以活性物種PdO為核心來發揮其最大性能,同時提高抗老化能力。這些發現為將來設計貴金屬催化劑提供了一定的參考。此外,Pt基催化劑性能還受到Pt粒徑的影響,Beck等[50]研究了一種簡便制備無氯單分散的Pt/Al2O3,并研究了粒徑與甲烷氧化活性之間的關系,如圖3(b)所示,發現平均粒徑在2 nm時達到了最佳性能,但在粒徑大于2 nm后活性逐漸下降并趨于穩定。
3.1.3 Au基催化劑
相較于Pd和Pt貴金屬元素,Au一直是被認為是惰性元素,應用較為狹窄。目前,提高Au基催化劑性能主要集中在Au性質和載體性質以及制備過程的優化。經研究Au與Pd/Pt之間的相互作用能夠有效提高催化活性。Miao等[51]探討了Pt的引入對于Au/Co3O4活性的影響,當加入少量Pt,Au/Co3O4對甲烷燃燒的活性明顯增強,完全氧化溫度下降了50 ℃,并且要優于Pt/Co3O4的性能;Wang等[52]將Au-Pd合金負載在3DOM La0.6Sr0.4MnO3載體上以探究3DOM負載貴金屬的優勢:大的比表面積和豐富的酸性位點有利于Au的高度分散和反應物的吸附和活化,并且貴金屬Au降低反應中間體和Pd結合強度,從而改變了甲烷氧化的反應途徑,與載體強烈的相互作用也有助于增強活性和穩定性;Han等[53]將該研究推進一步,探究載體上Co3O4組分對于AuPd/3DOM MnCo2O4影響,顯著增加了載體Co2+含量,從而導致高濃度吸附氧物種和氧空位,提高了整體催化活性。
3.2 負載型非貴金屬催化劑
貴金屬催化劑因其出色的低溫催化活性而被廣泛研究,并且已經開發出一系列的技術手段來降低高溫燒結等問題對于催化劑壽命和性能的影響,但不可否認的是,較差的熱穩定性和較高的成本依然限制著貴金屬催化劑大規模工業化應用。金屬氧化物在CH4催化燃燒領域同樣擁有較為廣泛應用環境,由于金屬氧化物當中的過渡金屬元素具備多重價態,易于在高氧化態和低氧化態之間形成氧化還原的閉合循環[54];同時,晶格氧的釋放和再次補充儲存導致了低溫條件下的高活性吸附氧以及高溫條件下的高活性晶格氧[55],這些條件造就了部分金屬氧化物催化活性可以與貴金屬催化劑相媲美。當然,與貴金屬相比,金屬氧化物的豐富儲量也使得其成本較為低廉,這也是近些年在研究和實際考慮中被偏向選擇的原因之一。金屬氧化物可以分為單一金屬氧化物和混合金屬氧化物(六鋁酸鹽、鈣鈦礦、尖晶石類),在這之中,Mn、Fe、Co、Ni、Cu等金屬氧化物催化劑成為研究的熱點。
3.2.1 CuO基催化劑
CuO是最為有效的CH4催化氧化材料之一,對其研究主要集中在將其負載在多孔載體上,因此,載體性質和Cu負載量是影響催化效果的主要因素[56,57]。Aguila等[58]評價了不同載體上單位質量CuO的催化活性。并得到以下順序:CuO/ZrO2>CuO/Al2O3≥ CuO/SiO2,表明了載體對CuO活性影響較大,其中,CuO/ZrO2較高活性于其表面上高度分散的Cu物種有關。Popescu等[59]探討了載體對于CuO催化劑的影響,比較了CuO在MgO、Al2O3和MgAlO載體上的催化性能。結果發現,在MgAlO載體上CuO活性物種從分布良好顆粒聚集成為CuAl2O4相,此時得到了最佳的催化活性;同時載體也影響到CuO基催化劑整體的表面酸堿性,CuO/MgAlO催化劑較高的強酸位點以及中、強堿性位點對于催化活性的提升起到了積極作用,壽命測試結果也展現了其在未來工業上的應用潛力。Park等[60]研究表明,孤立Cu表面相隨著Cu含量的增加而減少,并且提出孤立的Cu表面相比相互作用的Cu表面相或者結晶CuO對甲烷的氧化更為活躍。徐峰等[61]考察了Cu負載量對催化劑性能的影響,發現不同負載量對于Cu氧化態并無影響,且在負載量低于7%時,催化燃燒性能隨著負載量的增加而提高,但當其增加至10%時,由于CuO晶體顆粒變大,周圍電子云密度升高,活性物種分散性降低,導致了轉化率降低,催化性能下降。
3.2.2 Co3O4基催化劑
納米結構的Co3O4是一種典型尖晶石類過渡金屬氧化物,多孔結構、混合陽離子價態、豐富的活性氧種類以及良好的抗毒性和低成本效益等優勢使其在眾多過渡金屬氧化物催化劑中具有較高的吸引力[62]。研究表明,Co3O4的催化活性在一定程度上取決于其形態,這是由于不同晶面上的電子轉移特性各異,并且針對不同的被氧化物,所需要的優勢晶面也是不同的。對于CO,納米球狀以及棒狀Co3O4的氧化行為更為活躍,因為所包含的{110}上Co3+離子占主導地位,同時Co3+也被認為是CO氧化的活性位點[63,64];而對CH4燃燒有所不同,{001}和{111}平面上只有Co2+,這也被認為擁有著更多的氧空位,這對于CH4催化燃燒是極其有利的。Chen等[65]通過簡便的水熱法可控地合成了不同形態的Co3O4納米晶體,雖然對CH4燃燒都具有優異的催化活性,但是“花狀”和“六邊形板狀形”的Co3O4性能更為突出,這是由于該兩種形態更偏向于暴露出更多的活性{111}晶面,這就為針對Co3O4設計高活性的納米結構的金屬氧化物提供了重要啟示。Yu等[66]最近介紹了非金屬元素摻雜提升Co3O4活性的方法,其通過N2等離子體“雕刻”Co3O4方法實現雜原子N元素摻雜Co3O4催化氧化CH4的新方法,在反應溫度為342 ℃時實現了轉化率為6.5 μmol/g/s,約為原始催化劑的7.3倍,其優越的性能來自于結構缺陷和活性位點高度暴露,同時N元素摻雜提高了催化劑整體表面親電性,對Co3O4的活化起到了重要作用。該研究也為非金屬元素摻雜金屬氧化物提供了新思路。
Co3O4是CH4燃燒的良好催化劑,但是在550 ℃以上就會出現嚴重的燒結問題,明顯降低了其催化性能,可通過負載來增強Co離子穩定性。Feng等[67]利用熱穩定較強的SmMn2O5作為支撐,制備了Co3O4/SmMn2O5負載型催化劑,其中,Co3O4/SmMn2O5-50%樣品展現出最高的CH4燃燒催化特性,并且氧遷移速率增強。同時,與Co3O4相比,負載型催化劑的耐久性、熱穩定性以及催化壽命都得到了提高。
3.2.3 CeO2基催化劑
CeO2中的Ce陽離子十分活躍,能夠迅速從Ce4+轉化到Ce3+,過程伴隨著晶格氧的快速釋放,展現出良好的氧化還原性質,因此,基于CeO2制備的多相催化劑得到了廣泛的研究和應用。為了進一步提高CeO2基催化劑的儲氧能力,金屬陽離子摻雜這一方法經大量研究證明是有效的[68],所利用到的金屬陽離子主要包括Ba、Ca、Co、Cu、Mn、Nd、Pb、Sr、Y、Zn和Zr等。
Sánchez等[69]介紹了Ce0.5Tb0.5Ox/MgO新型催化劑,并與CeO2/MgO催化劑進行比較。結果發現,Ce0.5Tb0.5Ox/MgO在鑭系原子含量方面,Ce和Tb元素的總負荷量非常低,并且在低溫條件下具有高還原性以及高儲氧能力,比CeO2/MgO具有更高的催化活性,這源于Ce和Tb兩種鑭系元素在原子水平上的混合以及與MgO載體的相互作用。與CeO2相比,利用低于四價元素摻雜改善后的催化劑具有豐富的氧空位濃度,優越的氧空位性質以及獨特的氧儲存和氧化還原能力,此外,三價陽離子的引入比二價陽離子的引入更能增加氧的遷移率,這些都是基于Ce3+和Ce4+之間潛在的氧化還原過程當中優異的氧親和力和吸收、激發能帶所實現的[70]。Shan等[71]用不同方法制備了Ce1?xNixO2,結果表明,用溶膠凝膠法制備的Ce0.7Ni0.3O2顯示了最高的還原性和催化活性,且在Ce1?xNixO2催化劑中發現有三種類型的Ni相共存,即在CeO2載體上聚集的NiO、與CeO2有很強相互作用的NiO及融入CeO2晶格當中的Ni物種。Ce0.7Ni0.3O2最高催化活性不僅是歸因于高度分散的NiO物種,而在于形成的更活躍活性氧。
3.3 特殊結構氧化物催化劑
3.3.1 六鋁酸鹽型催化劑
六鋁酸鹽型催化劑具有極高的熱穩定性和抗燒結能力,自1987年以來,就被用于CH4氧化反應,因此,六鋁酸鹽型催化劑被認為是用于高溫條件最合適的催化劑。六鋁酸鹽型材料(AAl12O19?x,其中,A位是堿,堿土或者稀土陽離子)是一種六方層狀結構晶體,材料突出的熱穩定性主要與它們獨特的層狀結構有關,這些層狀結構由交替的Al2O3尖晶石塊組成,并被鏡面(其中含有大量的A位離子)隔離。根據鏡面層中陽離子的電荷、離子半徑和數目的不同可分為磁鉛石型和β-Al2O3型兩種類型[72,73]。迄今為止,大部分研究的關注焦點主要在選擇具有抗燒結性的金屬陽離子來取代六鋁酸鹽結構中的Al3+位離子,例如Cr、Mn、Fe、Co、Ni、Cu等,其中,Mn、Fe是過渡金屬中通過部分取代途徑促進六鋁酸鹽型催化劑活性的最佳備選元素[74]。
Yuranov等[75]通過對比各種陽離子取代的BaMAl11O19發現Mn取代的氧化物對甲烷的催化活性最高,這是由于Mn離子能夠在Mn2+和Mn3+價態之間的切換能力較強,但是過渡金屬的取代通常導致了六鋁酸鹽的比表面積下降,并且限制了氣體反應物的流動傳質。為了解決上述問題,Masato等[76]通過溶膠凝膠法-超臨界干燥法或反向微乳合成法合成了具有高活性和高比表面積(100 m2/g)的六氯酸鋇氧化物。雖然六鋁酸鹽型催化劑具有較高的熱穩定性和機械強度,但是與大部分催化劑相比,其催化活性較低,只能在CH4的多級催化中應用于最后一級;并且由于晶體結構部分取代程度較低,制備過程繁瑣,想以簡單便捷的方法工藝合成具備杰出催化性能的六鋁酸鹽型催化劑,還需要更進一步的研究。
3.3.2 尖晶石型催化劑
尖晶石氧化物屬于復合氧化物的一類,其化學通式為AB2O4,其中,A離子是占據了四面體位置的二價陽離子(例如Mg2+、Fe2+、Zn2+、Mn2+、Co2+),B離子是占據八面體位置的三價陽離子(例如Fe3+、Al3+、Cr3+、Mn3+、Co3+),并根據A、B離子的填充方式的改變和空間分布,可分為正尖晶石、反尖晶石、混合尖晶石三種類型[77],以NiCo2O4結構為代表,如圖4所示。
Mihai等[78]通過共沉淀法合成了MnCo2O4、NiCo2O4和CuCo2O4,用于CH4催化氧化過程,通過原位電導率測量來揭示在三種尖晶石上氧化機理。實驗結果發現,三種尖晶石分別遵循表面機制和界面機制。如圖5所示,其中,在接近催化實驗所用的條件下,MnCo2O4在Co3+還原為Co2+過程中,具備n型半導體的性質,當完全轉化為Co2+時產生氧空位;在Mn4+還原為Mn3+過程中,具備p型半導體性質,同時氣態氧吸附填充在氧空位處,整體氧化還原過程符合界面反應過程;而NiCo2O4和CuCo2O4在反應條件下,分別處于金屬導電狀態和p型半導體性質,其中,金屬陽離子都沒有發生氧化還原反應,說明在整個反應過程中表面機制發揮作用。
3.3.3 鈣鈦礦型催化劑
鈣鈦礦型氧化物(化學通用式為:ABO3)由交替的ABO3和AO層組成。周期表中絕大部分元素都可以參與鈣鈦礦的組成,并且可以以不同化合價態結合,例如:AⅠBⅤO3、AⅡBⅣO3、AⅢBⅢO3,正是由于多樣化的結合導致了鈣鈦礦結構的多重物化特性[15]。A位陽離子通常為堿金屬或者稀土元素,具有惰性的d0或者f0電子結構,主要作用是穩定整體結構,對氧化還原作用貢獻較小;B位離子通常是3d、4d、5d過渡金屬元素,但由于成本限制,一般使用3d電子結構元素,由于其具有進行逆向氧化還原循環而不會破壞結構的能力,所以成為結構中主要催化位點[79]。如圖6所示,在鈣鈦礦結構當中,較大的陽離子A位為12面體配位,而較小的陽離子B位為6面體配位,陽離子的半徑最低限制為RA> 0.09 nm和RB> 0.051 nm[80]。立方形結構是鈣鈦礦的理想結構,但是也存在一些具有差距的模型,例如三角對稱形,四邊形和正交菱形等,其結構存在限制界限,用容忍因子t來表達,一般鈣鈦礦的范圍為0.75 鈣鈦礦的微觀結構有利于氧遷移速率的加快以及催化活性的提升,這主要是由于鈣鈦礦氧化物催化燃燒CH4的活性氧為晶格氧(包括表面晶格氧和結構晶格氧),反應物吸附在材料表面與晶格氧進行反應后使整體結構缺陷,即生成氧空位,氧空位越多,在表面形成的吸附氧物種也就越多,符合MVK機理;當然,優異的離子遷移率和氧化還原性能以及熱穩定性也使得鈣鈦礦成為催化燃燒催化劑的選擇之一[20],但是目前鈣鈦礦的應用仍面臨著起燃溫度高,催化活性差等困難。為了優化鈣鈦礦物化性質,提高催化性能,目前,所使用到的改性技術如圖7所示,例如優化制備方法、A/B位陽離子部分或者雙取代等。 鈣鈦礦催化劑活性和穩定性與自身的性質有較大的關系,如粒徑分布、比表面積和結晶度等,由制備過程所決定,常用制備方法有:模板法、共沉淀法、溶膠凝膠法和溶液燃燒法等。Wang等[83]采用了共沉淀法、溶膠凝膠法(不同絡合劑)和模板法制備了LaCoO3來探究制備方法對于活性的影響。如圖8所示。 不同制備方法對于催化活性有明顯影響,其中,對于共沉淀法,由于前體中羥基析出物未發生分解因此鈣鈦礦結構并未形成,從而催化活性較差;對于溶膠凝膠法,采用單檸檬酸絡合劑時,鈣鈦礦整體微觀結構為顆粒狀,而采用乙二醇-甲醇雙絡合劑時,結構呈現多孔結構并形成較多氧空位,使得氧化還原性能增強;對于模板法,在模板煅燒過程中發生孔隙塌陷,所以未形成較好的多孔結構,其存在狀態為顆粒狀,但是該煅燒過程消耗了晶格氧,從而導致氧空位增加和更為活躍的晶格氧產生。該研究證明了鈣鈦礦的物化性質與表面形態、比表面積、表面成分組成以及氧化還原能力都與制備方法密切相關。然而,傳統制備方法(如溶膠凝膠法、共沉淀法等)都包括高溫固化過程,容易導致孔隙結構的破壞而限制了鈣鈦礦獲得更高的催化活性。王賽飛等[84]采用溶膠凝膠和水熱聯用法制備了La1?xSrxCoO3鈣鈦礦型催化劑,發現兩種制備手段聯用使得其大介孔含量明顯增加,比表面積明顯增加,呈現出多級孔協同作用,從而顯著減少積炭對于催化劑的影響,有效提高了對CH4的催化活性。 近年來,為了彌補傳統方法帶來的比表面積小的問題,三維有序大孔結構(3DOM: Threedimensionally ordered microporous)材料具備大的比表面積和孔容、規整及可控的孔徑、周期性的孔隙結構以及良好的傳質能力等優勢,因此,被廣泛研究和應用。3DOM材料的制備方法有刻蝕法(Etching method)、生 物 模 板 法(Biological template method)、膠體晶體模板法(Colloidal crystal template method),但前兩種方法需要特殊的設備、嚴格的條件和復雜的操作,所以降低了實驗室條件下的適用性;而膠體晶體模板法是一種簡便、成本低并且大部分條件下都具有適應性的方法,典型的操作流程如圖9所示[85]。Arandiyan等[86]利用膠體晶體模板法在P-123作為溶脹劑和介孔促成劑的條件下制備了伴有介孔壁的3DOM結構La1?xCexCoO3,與傳統La1?xCexCoO3相比,3DOM-La1?xCexCoO3對CH4具有較高的催化活性,這主要歸功于更高的比表面積和氧物種濃度、更好的低溫還原性以及獨特的孔隙結構。同時,Ce的加入明顯提高了熱穩定性。 對A/B位金屬陽離子進行摻雜或者取代可明顯增強鈣鈦礦催化活性,這是由于正常三價A位離子被二價/四價離子取代/摻雜后,會通過B位離子被氧化/還原或氧空位的產生來實現電荷補償;而對B位離子進行取代/摻雜會直接引起B位離子價態和活躍程度的改變,從而產生氧缺陷,提高表面活性氧物種濃度和晶格氧遷移速率,進而提高鈣鈦礦催化活性[87],該手段可以構造的摻雜/取代型結構包括A1?xA'xBO3、AB1?xB'xO3、A1?xA'xB1?xB'xO3(A = La、Nd、Sm,B = Fe、Co、Ni、Mn)。Santos等[88]利用檸檬酸絡合法制備了一系列的LaNi1?xCoxO3鈣鈦礦催化劑,同時利用原位XRD探究了在CH4氧化過程中LaNi0.5Co0.5O3和LaNi0.8Co0.2O3的結構變化,實驗發現Co的最佳替代度為x=0.5,并且根據材料的替代度,激活了不同的還原機制。如圖10所示,LaNi0.5Co0.5O3的還原過程涉及LaNi0.5Co0.5O2.5和Co3O4中間相的形成,而LaNi0.8Co0.2O3則顯示出了過氧化物、尖晶石相以及NiO相。雖然以Co代替Ni導致反應的活性氧減少。但是Co摻雜提高了催化劑的再氧化性,使表面焦炭沉積明顯下降。若當A = A' = B = B'時,此時就會出現一種新鈣鈦礦結構,即雙鈣鈦礦(AA'BB'O6),較單鈣鈦礦而言,由于其結構中多重金屬元素的相互作用,展現了其在催化反應中優勢。Gao等[89]采用簡便的溶膠凝膠法制備了CeO2/La2CoFeO6和La2CoFeO6/CeO2,并且CeO2/La2CoFeO6催化性能要優于La2CoFeO6/CeO2,這主要是因為CeO2和La2CoFeO6之間的相互作用和擁有豐富的吸附氧物種。 在優化制備過程和A/B位摻雜或者取代方法以外,利用溶液刻蝕鈣鈦礦結構和制備負載型鈣鈦礦,也是提高鈣鈦礦性能的另一途徑。Ding等[90]報道了一種通過有效而溫和的檸檬酸刻蝕LaMnO3催化劑來改進催化CH4燃燒活性,克服了之前利用強酸[91,92]刻蝕導致結構破壞,熱穩定性下降等缺點,刻蝕后的催化劑由于具有更高的比表面積,豐富的氧空位和更高的Mn4+/Mn3+,在保持整體結構的前提下催化活性得到了提升。該方法既調節了比表面積等物理性質,又改善了金屬離子價態比例等化學因素。而Jing等[93]通過制備La0.8Ca0.2FeO3/MgAl2O4,結構當中Ca和Mg部分交換使得鈣鈦礦的結構形貌和活性位點發生變化。同時該催化劑發揮雙重物相協同催化的作用,催化性能和熱穩定性顯著提高。 除了人為因素對鈣鈦礦催化劑性能影響外,工況當中的其他物質也會影響鈣鈦礦性能,其中,煤礦瓦斯處理轉化的關鍵問題是混合氣體中SO2和H2S的存在會導致催化劑中毒現象發生[94]。已有很多文獻對催化劑SO2中毒機制進行了研究,SO2吸附在催化劑表面后會迅速與活性氧發生反應,形成不穩定的表面物質,例如硫酸鹽、亞硫酸鹽或硫化物等,并且會從表面遷移到主體。根據中毒的程度,有時會使活性相完全破壞,形成分離的B位離子氧化物和硫酸A位離子鹽[95]。目前的解決辦法有摻雜貴金屬或過渡金屬氧化物等。Rosso等[96]制備了LaMn1?xMgxO3催化劑,能夠在800 ℃、2×10?4SO2環境中暴露32 d且催化活性無明顯影響,該結果表明,MgO物種的存在可以有效減緩催化劑的硫中毒。Lott等[97]探討了貴金屬Pd的添加對于LaCoO3和LaMnO3抗硫作用的影響,發現Pd粒子的加入提高了鈣鈦礦對于硫的吸附電位。另外Cr和Ce摻雜也可增強鈣鈦礦的抗硫性能[98,99]。 本綜述就CH4催化燃燒的催化機理和催化劑方面做出概述。通過對催化機理總結發現,“Twoterm”混合機制能隨著溫度變化充分結合表面吸附氧和結構中晶格氧兩種氧化途徑,相較于單一表面反應或界面反應機理,對催化燃燒過程解釋較為完整。針對于催化劑方面,貴金屬在低溫條件下具有優異的性能表現,然而易燒結等問題卻一直存在,雖通過負載等方法有所改善,但是高昂的成本使非貴金屬催化劑得到了開發和利用。通過對非貴金屬材料進行制備方法優化、金屬離子摻雜/取代、酸堿溶液刻蝕等改性工藝來改善材料表面的物化性質,有助于催化活性和抗毒性的提高。盡管對催化燃燒過程的反應機理和催化劑活性提升方面研究頗豐,但是目前催化劑的活性仍有提升的空間,不同催化劑上的反應機理仍需進一步明確。未來關于強化催化劑性能方面的研究重點可以側重于以下幾個方面: 第一,通過利用非金屬元素(例如:N、C、P等)活躍的電子分布來實現非金屬元素的結構摻雜,來避免金屬元素的過度利用,降低改性成本。 第二,通過優化催化劑結構和化學組分(例如:調節氧化物陽離子化學計量比或多物相負載等),進一步暴露活性位點和產生多種物相協同催化的效果。 第三,通過原位實驗和模擬計算來探究針對各類催化機理的準確和完整的描述符,從理論層面增強實驗研究的方向性和針對性。4 結 語
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
現代企業(2015年9期)2015-02-28 18:56:50

- 燃料化學學報的其它文章
- Bi含量對溴氧化鉍光催化性能的影響
- Nickel oxide modified C3N5 photocatalyst for enhanced hydrogen evolution performance
- 催化裂化反應對1-己烯疊合反應的影響規律
- Phosphorous modified V-MCM-41 catalysts for propane dehydrogenation
- γ-Fe2O3納米顆粒尺寸及碳化氣氛對碳化過程的影響
- Methanol converting to propylene on weakly acidic and hierarchical porous MFI zeolite