分子模擬技術(shù)在高嶺石研究中的應(yīng)用進(jìn)展
2022-02-22 09:58:28楊有威羅玉霞張青青王春英
硅酸鹽通報(bào) 2022年1期
楊有威,羅玉霞,張青青,王春英
(江西省礦冶環(huán)境污染控制重點(diǎn)實(shí)驗(yàn)室,贛州 341000)
0 引 言
高嶺石是高嶺土的主要成分[1],得名于江西省景德鎮(zhèn)的高嶺山,其具有土狀光澤,呈白色或灰白色,分子式為Al4[Si4O10](OH)8,化學(xué)組分為39.50%(質(zhì)量分?jǐn)?shù))AlO3,46.54%(質(zhì)量分?jǐn)?shù))SiO2,13.96%(質(zhì)量分?jǐn)?shù))H2O,屬三斜晶系。硅氧四面體(SiO4)和鋁氧八面體(AlO6)靠O原子連接成1 ∶1型層狀結(jié)構(gòu),層間的O原子形成氫鍵,從而構(gòu)成了重疊的高嶺石層狀分子[2-3]。作為典型的鋁硅酸鹽層狀黏土礦物[4],高嶺石具有良好的電絕緣性、耐火性、可塑性及一定的白亮度,因此在陶瓷、涂料、催化劑、吸附劑等領(lǐng)域具有廣泛的應(yīng)用[5-9]。隨著21世紀(jì)科學(xué)技術(shù)不斷發(fā)展,分子模擬技術(shù)作為一種基于經(jīng)典力學(xué)和量子力學(xué)等理論的計(jì)算模擬方法已經(jīng)成為研究物質(zhì)分子水平性質(zhì)的有力工具,應(yīng)用十分廣泛[10-13]。
1 分子模擬技術(shù)及方法
分子模擬技術(shù)是一種基于PC端的計(jì)算模擬方法,通過(guò)運(yùn)用一些模擬軟件,將試驗(yàn)得到的原始結(jié)構(gòu)數(shù)據(jù)化,將數(shù)據(jù)導(dǎo)入軟件構(gòu)建物質(zhì)模型,驗(yàn)證其合理性后確定物質(zhì)微觀結(jié)構(gòu)。分子模擬不僅可以在分子層面模擬物質(zhì)的結(jié)構(gòu),還能夠模擬物質(zhì)發(fā)生反應(yīng)前后分子的運(yùn)動(dòng)變化,這使其在科學(xué)研究中的作用愈加明顯[14]。
目前使用較為廣泛的分子模擬軟件有Nanoscale Molecular Dynamics(NAMD)、Vienna Ab-initio Simulation Package(VASP)和Materials Studio(MS)等。NAMD是在計(jì)算機(jī)上快速模擬大分子體系并進(jìn)行動(dòng)力學(xué)模擬的代碼包,使用經(jīng)驗(yàn)力場(chǎng),通過(guò)數(shù)值求解運(yùn)動(dòng)方程計(jì)算原子軌跡。NAMD是眾多計(jì)算模擬軟件中并行處理最好的,可以支持幾千個(gè)CPU運(yùn)算,模擬體系原子數(shù)可達(dá)103~106個(gè),適合模擬蛋白質(zhì)、核酸、細(xì)胞膜等體系。VASP是維也納大學(xué)Hafner小組開(kāi)發(fā)的運(yùn)用平面波贗勢(shì)方法進(jìn)行電子結(jié)構(gòu)計(jì)算和量子力學(xué)-分子動(dòng)力學(xué)模擬的軟件包,通過(guò)近似求解Schr?dinger方程得到體系的電子態(tài)和能量,既可以在密度泛函理論(DFT)框架內(nèi)求解Kohn-Sham方程,也可以在Hartree-Fock(HF)的近似下求解Roothaan方程;VASP可以自動(dòng)確定任意構(gòu)型的對(duì)稱(chēng)性,利用對(duì)稱(chēng)性可設(shè)定Monkhorst-Pack特殊點(diǎn),便于高效計(jì)算體材料和對(duì)稱(chēng)團(tuán)簇,不過(guò),軟件運(yùn)行基于 Linux 操作系統(tǒng),操作不及MS方便。MS是美國(guó)Accelrys公司在2000年專(zhuān)門(mén)為材料科學(xué)領(lǐng)域研究設(shè)計(jì)的一款PC端運(yùn)行的模擬軟件,能夠構(gòu)建分子、固體及表面等結(jié)構(gòu)模型,通過(guò)運(yùn)用第一性原理近似求解薛定諤方程,預(yù)測(cè)材料的物理化學(xué)性質(zhì),以及模擬催化、聚合等化學(xué)反應(yīng)。MS在Windows、Linux 操作系統(tǒng)中均可運(yùn)行,界面友好,包含4大板塊23個(gè)模塊,實(shí)用方便,但其開(kāi)放性不如VASP,且并行效率不高。
分子模擬方法主要包括量子力學(xué)和經(jīng)典力學(xué)。量子力學(xué)模擬方法包括以DFT為依據(jù)的第一性原理計(jì)算法、半經(jīng)驗(yàn)法(Semi-enpirical)和從頭算法(Ab initial)。經(jīng)典力學(xué)模擬方法主要有分子力學(xué)方法(MM)、分子動(dòng)力學(xué)方法(MD)和蒙特卡羅方法(MC)等。高嶺石分子模擬研究中,使用的方法有第一性原理計(jì)算法、分子力學(xué)法、分子動(dòng)力學(xué)法和蒙特卡羅方法[15-19]。
第一性原理計(jì)算法根據(jù)軌道近似、非相對(duì)論近似和玻恩近似建立計(jì)算模型,并對(duì)薛定諤方程作近似處理,只需基本物理量就能用從頭算法進(jìn)行模擬計(jì)算,無(wú)需任何經(jīng)驗(yàn)參數(shù)對(duì)物質(zhì)體系的性質(zhì)和結(jié)構(gòu)進(jìn)行預(yù)測(cè)和分析,這種計(jì)算結(jié)果比半經(jīng)驗(yàn)法應(yīng)用程度更好。通過(guò)將多粒子轉(zhuǎn)化成多電子的量子力學(xué)方法,幫助解決了許多難以解釋的物理化學(xué)問(wèn)題[20]。
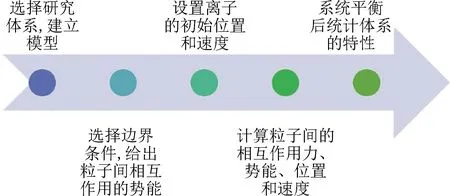
圖1 分子動(dòng)力學(xué)模擬流程圖[22]Fig.1 Molecular dynamics simulation flow chart[22]
分子動(dòng)力學(xué)方法是一門(mén)將數(shù)學(xué)、化學(xué)和物理結(jié)合成一體的方法。以牛頓第二定律為基礎(chǔ),描述模擬分子體系的運(yùn)動(dòng)變化,從系統(tǒng)中抽取樣本計(jì)算構(gòu)型函數(shù),求解[21]得到模擬體系中原子或分子的位移、速度、加速度等數(shù)據(jù)。通過(guò)對(duì)系統(tǒng)內(nèi)分子運(yùn)動(dòng)軌跡進(jìn)行分析處理,可以得到粒子的徑向分布函數(shù)、均方位移以及自擴(kuò)散系數(shù)等,隨后利用得到的數(shù)據(jù)對(duì)粒子的性質(zhì)進(jìn)行分析,分子動(dòng)力學(xué)模擬方法的主要流程[22]見(jiàn)圖1。
其中,初始設(shè)置的位置和速度是隨機(jī)選擇的,依靠溫度和速度大小進(jìn)行校正,校正結(jié)果保證了體系總動(dòng)量(P)為零(見(jiàn)式(1))。
(1)
式中:mi為第i個(gè)原子的質(zhì)量;vi為第i個(gè)原子的速度;N為體系原子數(shù)。
由于Bolzmann分布隨機(jī)選取速度為vi的第i個(gè)原子,當(dāng)溫度為T(mén)時(shí),原子在X軸上速度vix的概率密度ρ(vix)為:
(2)
其中一定速度下體系的溫度(瞬時(shí))可通過(guò)公式(3)求得:
(3)
式中:N為體系原子數(shù);mi為第i個(gè)原子的質(zhì)量,g;Pi為第i個(gè)原子的動(dòng)量,J;kB為Bolzmann常數(shù),1.380 66×10-23J/K。
分子力學(xué)模擬方法是根據(jù)經(jīng)典力學(xué)中的分子力場(chǎng)進(jìn)行計(jì)算模擬的。計(jì)算時(shí)有三個(gè)基于核間的近似假設(shè):(1)不考慮電子本身運(yùn)動(dòng)和分子力場(chǎng)參數(shù)的基礎(chǔ)上,根據(jù)某一個(gè)原子的原子核所在位置,即波恩-奧本海默近似;(2)分子是通過(guò)化學(xué)鍵作用聚集起來(lái)的原子團(tuán);(3)分子的基本單元在不同的分子中仍然具有結(jié)構(gòu)上的相似性。只考慮分子中化學(xué)鍵的伸縮、旋轉(zhuǎn)和鍵角的變化,通過(guò)能量數(shù)值描述變化,尋找合適力場(chǎng)中勢(shì)能最低的穩(wěn)定構(gòu)型,其中力場(chǎng)選擇的正確與否決定了計(jì)算結(jié)果是否可靠。
蒙特卡羅方法是一種基于熱力學(xué)進(jìn)行隨機(jī)抽樣的統(tǒng)計(jì)計(jì)算方法,抽樣是蒙特卡羅方法的核心原理。在系統(tǒng)條件下,采用Metropolis抽樣方法,生成微觀粒子隨機(jī)構(gòu)型,Boltzmann分布逐漸趨近于平衡后,根據(jù)給定的分子位能函數(shù),將粒子間內(nèi)能加和,得到能量數(shù)據(jù)。具體計(jì)算中每產(chǎn)生一個(gè)隨機(jī)狀態(tài),粒子都包含3種可能的操作:粒子的插入、刪除和移動(dòng)。
(1)粒子插入。在體系中的隨機(jī)位置插入一個(gè)粒子,概率為:
(4)
(2)粒子刪除。在體系中隨機(jī)刪除一個(gè)粒子,概率為:
(5)
(3)粒子移動(dòng)。在體系中隨機(jī)選取一個(gè)粒子移動(dòng)到另一位置,概率為:
pmove(s→s′)=min(1,exp{-β[U(s′N(xiāo))-U(sN)]})
(6)
式中:U為構(gòu)型總勢(shì)能,J/mol;V為體系體積,m3;N為粒子數(shù);μ為化學(xué)勢(shì),J/mol;?為德布羅意波長(zhǎng),m;β=1/kBT;s和s′為移動(dòng)前后粒子在體系中的狀態(tài)。
2 分子模擬方法在高嶺石研究中的應(yīng)用進(jìn)展
2.1 高嶺石晶體表面結(jié)構(gòu)特性的模擬研究
Gruner[23]于1932年采用X射線粉末衍射儀對(duì)高嶺石表面性質(zhì)進(jìn)行了分析,發(fā)現(xiàn)高嶺石晶胞空間群為Cc,屬于包含兩個(gè)結(jié)構(gòu)層的單斜晶系。此后,Bish[24]在1993年研究得到了高嶺石結(jié)構(gòu)參數(shù)(a=0.515 35 nm,b=0.894 19 nm,c=0.739 06 nm,α=91.926°,β=105.046°,γ=89.797°),空間結(jié)群C1最為準(zhǔn)確,提出了高嶺石屬三斜晶系的觀點(diǎn),具體顯示見(jiàn)圖2,此參數(shù)目前被作為研究高嶺石的基本標(biāo)準(zhǔn),并得到廣泛的運(yùn)用。高嶺石是多晶面礦物,李海普等[25]研究表明,高嶺石在(001)面發(fā)生的解離只是結(jié)構(gòu)單元層間氫鍵的斷裂,沒(méi)有化學(xué)鍵的斷裂,并且在自然破碎下(001)面的解離相比于其他晶面更加完全,事實(shí)上高嶺石的(001)面在高嶺石顆粒總面積中所占比例也是最大的。胡雪飛[26]基于密度泛函理論對(duì)高嶺石進(jìn)行研究,證明高嶺石(001)面較易解離。首先運(yùn)用MS軟件CASTEP模塊對(duì)高嶺石晶體模型進(jìn)行優(yōu)化,通過(guò)收斂性測(cè)試確定了模擬的構(gòu)型參數(shù),在此基礎(chǔ)下,選用Buid功能建立超胞模型改用CASTEP模塊Task任務(wù)優(yōu)化后計(jì)算高嶺石晶體結(jié)構(gòu)的能帶結(jié)構(gòu)、電荷布居、態(tài)密度等性質(zhì),對(duì)比后發(fā)現(xiàn)O原子與Si原子之間的作用強(qiáng)度更強(qiáng),高嶺石晶體易沿著(001)面解離。無(wú)論是理論模擬還是實(shí)驗(yàn)測(cè)試,均已對(duì)高嶺石的結(jié)構(gòu)特性有了充分研究,目前其結(jié)構(gòu)特性已不是研究重點(diǎn)。
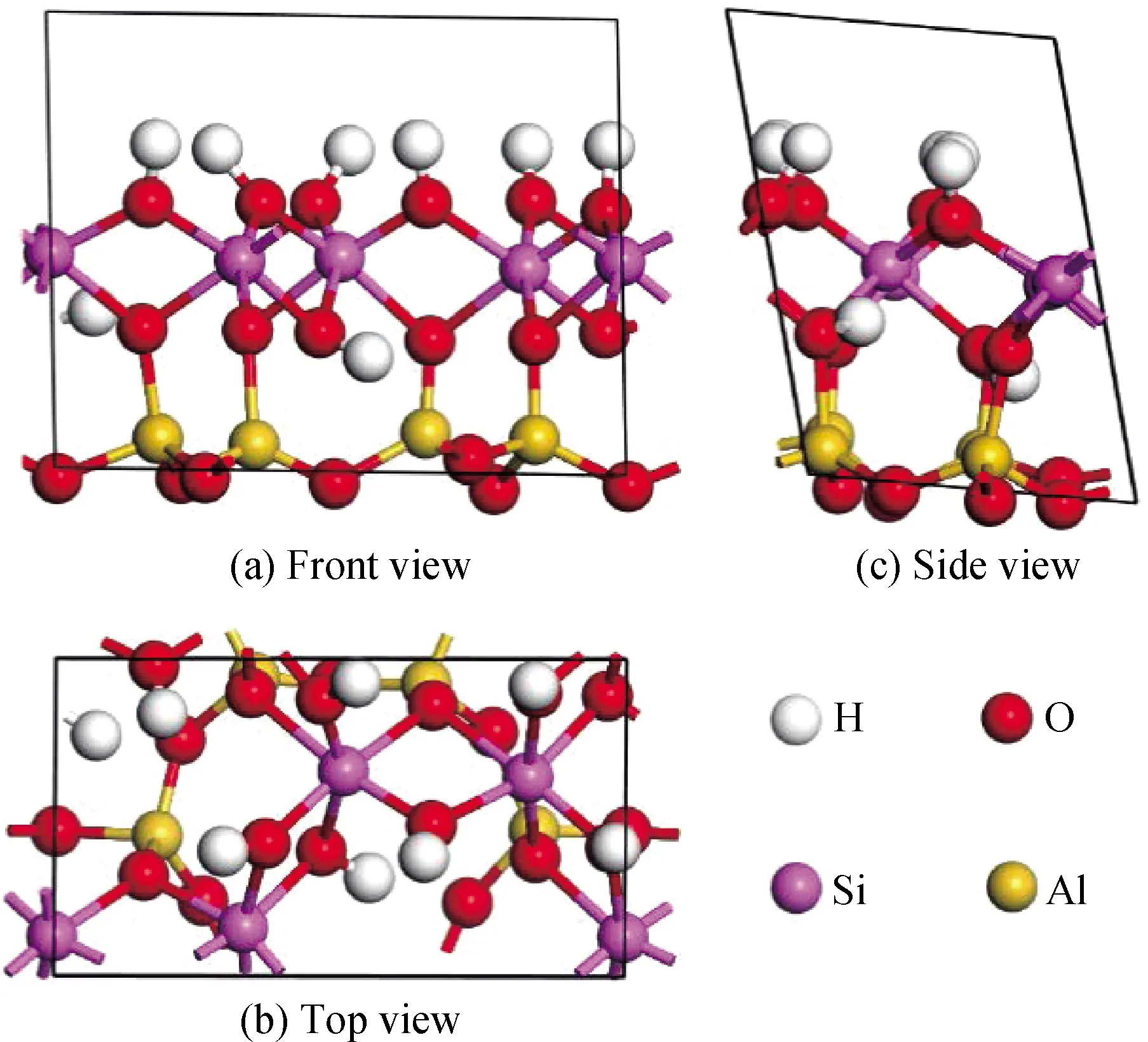
圖2 高嶺石初始構(gòu)型三視圖Fig.2 Three views of initial configuration of kaolinite
2.2 高嶺石改性的模擬研究
目前,已有多個(gè)研究證明了高嶺石對(duì)環(huán)境污染物的吸附能力[27-29]。對(duì)于高嶺石的吸附原理研究也在不斷進(jìn)行,韓永華等[30]對(duì)羥基鈣在高嶺石表面的吸附機(jī)理進(jìn)行了探索,采用MS軟件的CASTEP模塊優(yōu)化,以285 eV的截?cái)嗄堋DA(local density approximation)函數(shù)和4×2×3的K點(diǎn)為計(jì)算條件,優(yōu)化后建立起2×2×1超胞模型,改用Dmol3模塊對(duì)已優(yōu)化的表面模型及Ca(OH)+進(jìn)行能量計(jì)算。結(jié)果表明,羥基鈣吸附可在硅氧面和鋁氧面進(jìn)行,在鋁氧面氫原子的1s軌道與羥基鈣中氧原子的2p軌道雜化成鍵,在硅氧面兩者以靜電吸附的方式結(jié)合。硅氧面的吸附較為穩(wěn)定,水分子會(huì)影響鋁氧面的吸附,說(shuō)明高嶺石雖不是水化膨脹類(lèi)黏土礦物,但水分子的存在可以影響高嶺石對(duì)物質(zhì)的吸附。分子模擬技術(shù)的另一特點(diǎn)是可以較易滿(mǎn)足試驗(yàn)研究所需的條件,如對(duì)高嶺石進(jìn)行改性摻雜[31]。Zhang等[32]首先合成了煤矸石/g-C3N4催化劑活化過(guò)硫酸鹽,采用第一性原理計(jì)算得出煤矸石中的高嶺石降低了過(guò)硫酸鹽的吸附能,與復(fù)合改性前相比,合成的催化劑可以有效地吸附和活化過(guò)硫酸鹽產(chǎn)生的活性自由基,與試驗(yàn)結(jié)果一致。Scholtzov等[33]對(duì)高嶺石差層復(fù)合改性進(jìn)行了研究,通過(guò)第一性原理模擬計(jì)算了純高嶺石、甲醇插層高嶺石、甲氧基接枝高嶺石、混合接枝/插層高嶺石和含水混合高嶺石的彈性常數(shù)、體積、剪切和楊氏模量等力學(xué)參數(shù),分析得到了高嶺石最穩(wěn)定的改性方法為接枝/插層。周麗萍[34]通過(guò)CASTEP模塊優(yōu)化被摻雜高嶺石,優(yōu)化后的高嶺石表面電荷分布、態(tài)密度和布居分析結(jié)果顯示高嶺石表面的Al原子可以被Fe/Mg/Ca原子單取代或雙取代(見(jiàn)圖3),從而使高嶺石表面具有更多的負(fù)電荷,增強(qiáng)了表面共價(jià)性,從而增加了成鍵可能性。同時(shí)以水合鉛離子作為吸附質(zhì),研究其在Fe/Mg/Ca摻雜高嶺石表面的吸附,與未摻雜的高嶺石對(duì)比,證實(shí)了可以通過(guò)摻雜改性提高高嶺石的吸附性能。實(shí)際上,Rybka等[35]合成了零價(jià)鐵修飾改性高嶺石,并將其用作水溶液中Pb(II)和Mo(VI)的吸附劑,試驗(yàn)結(jié)果表明,合成的改性高嶺石對(duì)Pb(II)和Mo(VI)的吸附量相對(duì)于未改性高嶺石有顯著提高,印證了高嶺石可通過(guò)改性方式提高吸附特性。
運(yùn)用密度泛函/第一性原理進(jìn)行理論計(jì)算時(shí),多采用CASTEP模塊和Dmol3模塊(以MS軟件為例),對(duì)于能量計(jì)算,一般Dmol3模塊更為準(zhǔn)確,具體計(jì)算步驟為,CASTEP模塊/Dmol3模塊對(duì)初始單胞進(jìn)行結(jié)構(gòu)優(yōu)化,由Buid模塊建立合適的超胞模型并優(yōu)化,然后進(jìn)行過(guò)渡態(tài)搜索或吸附能計(jì)算,最后通過(guò)模塊自帶的分析功能選擇分析數(shù)據(jù),計(jì)算參數(shù)一般取自經(jīng)驗(yàn)參數(shù)。而運(yùn)用分子力學(xué)和分子動(dòng)力學(xué)原理計(jì)算時(shí)一般以Forcite模塊和Adsorption Locator模塊為主,不同的是建立超胞模型后可設(shè)置溫度、壓強(qiáng)和孔隙率等參數(shù)對(duì)模型的淬火、退火、競(jìng)爭(zhēng)吸附等行為進(jìn)行模擬,分子變化能夠很直觀地在界面體現(xiàn),具體數(shù)據(jù)可在返還的文件中找到或在模塊分析中導(dǎo)出。
近年來(lái)的研究表明,基于密度泛函理論,分子模擬技術(shù)的應(yīng)用對(duì)高嶺石的開(kāi)發(fā)研究具有理論指導(dǎo)意義,不但完善了高嶺石反應(yīng)機(jī)理,還為高嶺石提供了改性方向和方法。基于此,未來(lái)的模擬技術(shù)應(yīng)用可將其定位為對(duì)高嶺石變化特征的一種微觀研究方法,例如元素插入對(duì)高嶺石的影響或高嶺石與其他材料復(fù)合后的變化等,使高嶺石材料的研究更富有說(shuō)服力和可信度。
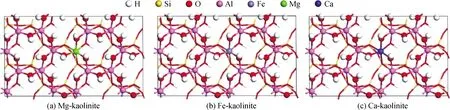
圖3 對(duì)高嶺石初始結(jié)構(gòu)擴(kuò)展2×2×1Fig.3 Initial structure of kaolinite expanded by 2×2×1
2.3 高嶺石對(duì)離子/分子吸附的模擬研究
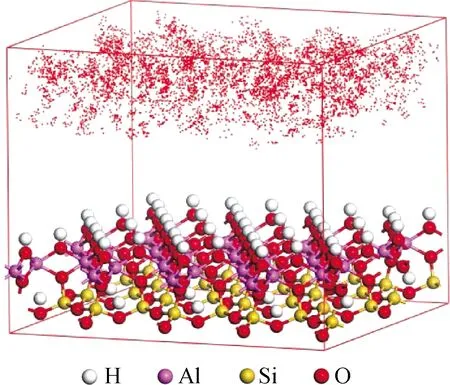
圖4 高嶺石吸附Pb2+密度分布Fig.4 Density distribution of Pb2+ adsorbed by kaolinite
已有實(shí)驗(yàn)證明高嶺石與多數(shù)離子相互吸附[36],但離子與高嶺石之間的相互作用機(jī)理仍需分子層面的理論解釋。Chen等[37]通過(guò)經(jīng)典分子動(dòng)力學(xué)模擬研究了Cs+在高嶺石中的吸附和擴(kuò)散行為,結(jié)果表明,Cs+優(yōu)先以表面絡(luò)合物的形式吸附在硅氧面,并且隨著溫度的升高,Cs+的擴(kuò)散系數(shù)顯著增加。然而,不同的高嶺石濃度和競(jìng)爭(zhēng)陽(yáng)離子對(duì)Cs+的擴(kuò)散系數(shù)會(huì)有輕微影響;Zhu等[38]采用分子動(dòng)力學(xué)模擬研究了磺基水楊酸與鋁、稀土離子(鑭和釔)的相互作用以及在高嶺石表面的吸附,結(jié)果表明,磺基水楊酸和鋁之間發(fā)生絡(luò)合反應(yīng),形成O—Al共價(jià)鍵,但與稀土離子之間只有弱吸附,因此,磺基水楊酸可以與游離鋁離子形成絡(luò)合物,也可以通過(guò)與高嶺石(100)面的鋁形成氫鍵吸附在高嶺石上,最后通過(guò)XPS和SEM的檢測(cè)和分析,驗(yàn)證了分子動(dòng)力學(xué)模擬結(jié)果;Zhang等[39]用密度泛函理論計(jì)算和蒙特卡羅方法研究了鈣和鉀在高嶺石表面的微觀吸附機(jī)理,得到了鈣離子和鉀離子在高嶺石(001)和(001)面上的吸附結(jié)構(gòu),計(jì)算了其結(jié)合能,結(jié)果顯示吸附在高嶺石(001)面的主要產(chǎn)物為鈣(II)和鉀(I),結(jié)合穆利肯鍵電荷和布居數(shù)的部分態(tài)密度投影表明,吸附產(chǎn)物的鈣氧鍵和鉀氧鍵受離子相互作用支配。陽(yáng)離子水合可以增加吸附發(fā)生的概率,增強(qiáng)鍵間的共價(jià)相互作用,之后的吸附實(shí)驗(yàn)證明鈣離子比鉀離子更容易吸附在高嶺石上;Chen等[40]通過(guò)第一性原理計(jì)算探討了鎘在高嶺石基面上的吸附機(jī)理。結(jié)果表明,鎘(II)在Kln-Al(001)面的雙齒配位優(yōu)于所有其他研究模型,計(jì)算的電子密度差揭示了吸附時(shí)表面氧向鎘電荷轉(zhuǎn)移,部分態(tài)密度分析顯示,由于Cd-5p和Os-2p軌道在價(jià)帶中的重疊,Cd-Os鍵表現(xiàn)出共價(jià)特征。此外,利用AIMD模擬的徑向分布函數(shù)確定了高嶺石-水界面鎘配位殼的結(jié)構(gòu)特征。楊飛等[41]研究了不同溫度下高嶺石對(duì)重金屬離子的吸附,研究表明,在288 K到308 K區(qū)間內(nèi),隨著溫度的升高高嶺石對(duì)Pb2+、Cd2+的吸附量逐漸降低,密度分布見(jiàn)圖4,從吸附量上看,高嶺石對(duì)吸附Pb2+更有利,且在兩種離子的競(jìng)爭(zhēng)吸附中Pb2+強(qiáng)于Cd2+,吸附結(jié)果與Sari等[42]和Zhong等[43]的實(shí)驗(yàn)測(cè)試結(jié)果基本吻合。
分子模擬技術(shù)目前應(yīng)用于解釋高嶺石對(duì)離子/分子的吸附機(jī)理,對(duì)于實(shí)驗(yàn)中高嶺石去除污染物的現(xiàn)象,直觀模型能很好地展示高嶺石吸附前后的變化。分子模擬方法已經(jīng)成為研究黏土礦物和其他層狀結(jié)構(gòu)材料孔隙中離子/分子能量學(xué)、結(jié)構(gòu)和動(dòng)力學(xué)的有效工具,為實(shí)驗(yàn)結(jié)果提供了有價(jià)值的補(bǔ)充和指導(dǎo),是高嶺石吸附研究中應(yīng)用較為廣泛的方法之一,未來(lái)應(yīng)用重點(diǎn)依然是對(duì)吸附機(jī)理的微觀變化解釋。
2.4 高嶺石在選礦/采礦領(lǐng)域的模擬研究
區(qū)別于密度泛函理論研究的吸附,分子動(dòng)力學(xué)、分子力學(xué)和蒙特卡洛方法多組合用于研究高嶺石礦物吸附目標(biāo)礦產(chǎn)/浮選藥劑性能。在選礦/采礦(主要是采氣)領(lǐng)域, Ma等[44]對(duì)多種物質(zhì)分子在高嶺石表面的吸附行為進(jìn)行了模擬研究,結(jié)果表明,溫度和壓力對(duì)高嶺石(001)面吸附特性有重要影響。在較低溫度條件下,CO2在高嶺石表面的吸附距離小于CH4,但在溫度為373 K、壓力為25 MPa的條件下,CO2在高嶺石表面的吸附距離大于CH4。氟碳表面活性劑在高嶺石表面的吸附距離遠(yuǎn)小于H2O、CO2、CH4、N2和C8H18體系,主要是氟碳表面活性劑與高嶺石表面具有較強(qiáng)的氫鍵相互作用,可以改變高嶺石表面的潤(rùn)濕性,不僅提高了油氣采收率,還削弱了儲(chǔ)層親水性,減弱了高嶺石孔隙的鎖水效應(yīng)。Liu等[45]和唐巨鵬等[46]還研究了高嶺石對(duì)頁(yè)巖氣的吸附性能規(guī)律,由于頁(yè)巖氣主要存在于頁(yè)巖層中的有機(jī)質(zhì)和黏土礦物中,占到了總氣量的20%~85%(體積分?jǐn)?shù))[47],因此,兩位學(xué)者對(duì)高嶺石吸附頁(yè)巖氣主要成分CH4進(jìn)行了研究,蒙特卡洛和分子動(dòng)力學(xué)研究表明,高嶺石吸附CH4時(shí)優(yōu)先發(fā)生在硅氧面上,為物理吸附,但高溫不利于其吸附CH4分子,含水率的增加會(huì)減少其對(duì)CH4的吸附量,研究結(jié)果為頁(yè)巖開(kāi)采提供了一定的理論支持。Han等[48]研究六偏磷酸鈉在高嶺石顆粒上的分散機(jī)理(六偏磷酸鈉是浮選工藝和黏土工業(yè)中廣泛使用的分散劑),應(yīng)用分子動(dòng)力學(xué)和密度泛函理論模擬了線性聚磷酸鹽鏈與高嶺石鋁羥基封端表面的相互作用及 [HPO4]2-與高嶺石鋁羥基表面的相互作用,解釋了高嶺石與分散劑之間的吸附機(jī)理。黃藥作為選礦藥劑廣泛應(yīng)用于選礦,Zhang等[49]考慮到高嶺石是采礦中常見(jiàn)的脈石礦物,有必要了解其與黃藥的相互作用,因此通過(guò)表征手段、分子動(dòng)力學(xué)模擬和密度函數(shù)理論研究了黃藥和高嶺土表面的相互作用。結(jié)果表明,黃藥在高嶺土表面的吸附符合偽一級(jí)(PFO)和朗格繆爾模型,黃藥分子以單層吸附在高嶺土表面,影響了廢水處理過(guò)程中高嶺土和絮凝劑之間的相互作用。Chang等[50]采用實(shí)驗(yàn)和分子動(dòng)力學(xué)模擬方法,綜合研究了聚丙烯酰胺對(duì)鋁土礦浮選的影響,發(fā)現(xiàn)當(dāng)聚丙烯酰胺濃度超過(guò)臨界值時(shí),鋁土礦浮選的回收率和選擇性迅速下降。研究表明,聚丙烯酰胺可以通過(guò)其酰胺基與礦物表面的氫氧化鋁之間的氫鍵作用吸附在一水硬鋁石和高嶺石上。低濃度時(shí),聚丙烯酰胺的烴鏈向外取向,增加了礦物表面的疏水性,而高濃度時(shí),聚丙烯酰胺分子通過(guò)分子間作用相互纏結(jié),降低了礦物表面的疏水性,結(jié)果與接觸角測(cè)量結(jié)果吻合較好。Ziemiański等[51]通過(guò)高壓和低壓氣體吸附技術(shù)研究了黏土礦物吸附甲烷的影響因素,研究表明,黏土礦物的溫度、含水量、陽(yáng)離子和孔隙壓力等因素都會(huì)影響吸附,吸附位點(diǎn)主要位于黏土礦物的層間距表面,不同類(lèi)型的黏土礦物是控制甲烷吸附的主要因素,佐證了高嶺石吸附甲烷的模擬研究結(jié)果。
綜合來(lái)看,應(yīng)用分子模擬技術(shù)研究與高嶺石相關(guān)的選礦/浮選領(lǐng)域報(bào)道甚廣。高嶺石作為黏土礦物的主要成分之一,在許多礦產(chǎn)中均能發(fā)現(xiàn)高嶺石的存在。因此,高嶺石與礦產(chǎn)之間的吸附機(jī)理,與浮選藥劑之間的作用機(jī)理成為研究熱點(diǎn)。基于礦產(chǎn)資源高效開(kāi)發(fā)利用,未來(lái)研究的主要方向依然是與選礦/浮選有關(guān)的高嶺石改性吸附。
3 結(jié)語(yǔ)與展望
目前,高嶺石吸附離子/分子的模擬研究已從多方面展開(kāi)。近年礦產(chǎn)資源開(kāi)發(fā)利用方面應(yīng)用模擬技術(shù)進(jìn)行的研究更偏向于外部環(huán)境因素(物理、化學(xué))對(duì)高嶺石晶體構(gòu)型變化規(guī)律和對(duì)物質(zhì)吸附特性的影響,如水含量和空隙壓力對(duì)高嶺石吸附CH4的影響或溫度、金屬離子的存在是否會(huì)引起高嶺石表面的水化等。礦產(chǎn)資源開(kāi)發(fā)利用中常能看到高嶺石的影子,許多的采礦/浮選實(shí)驗(yàn)還留有空白,需要理論計(jì)算填補(bǔ)。在已有的改性高嶺石分子模擬中,國(guó)內(nèi)外對(duì)其晶體結(jié)構(gòu)和吸附特性的相關(guān)性尚未給出有力證據(jù)。因而,在實(shí)驗(yàn)基礎(chǔ)上建立分子模擬研究模型,有利于深入探討改性高嶺石晶體結(jié)構(gòu)和吸附特性的相關(guān)性。
近幾年分子模擬技術(shù)作為熱門(mén)的新興研究方法,廣泛應(yīng)用在材料研究領(lǐng)域。分子模擬技術(shù)具有指導(dǎo)實(shí)驗(yàn)方向,不受實(shí)驗(yàn)條件限制,最大限度地避免人為實(shí)驗(yàn)所帶來(lái)的誤差等優(yōu)點(diǎn)。
綜合近年國(guó)內(nèi)外學(xué)者的研究,在高嶺石開(kāi)發(fā)研究的應(yīng)用中,國(guó)內(nèi)的計(jì)算模擬研究要少于國(guó)外,對(duì)實(shí)驗(yàn)現(xiàn)象的描述以各類(lèi)表征手段為主。因此,國(guó)內(nèi)學(xué)者無(wú)論是在高嶺石吸附、改性或是在礦產(chǎn)開(kāi)發(fā)研究中都可將分子模擬技術(shù)作為一種可應(yīng)用的表征手段,以填補(bǔ)理論計(jì)算的空白。未來(lái)高嶺石的研究路線應(yīng)趨向于理論模擬計(jì)算指導(dǎo)實(shí)驗(yàn),實(shí)驗(yàn)驗(yàn)證模擬計(jì)算結(jié)果。隨著模擬方法和實(shí)驗(yàn)表征技術(shù)的不斷發(fā)展,分子模擬技術(shù)在物質(zhì)性質(zhì)研究及開(kāi)發(fā)過(guò)程中將發(fā)揮重要作用。
猜你喜歡
體育科技文獻(xiàn)通報(bào)(2022年3期)2022-05-23 13:46:54
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機(jī)設(shè)計(jì)與研究(2019年4期)2019-05-21 07:21:24
中國(guó)塑料(2016年12期)2016-06-15 20:30:07
Coco薇(2016年2期)2016-03-22 02:42:52
中國(guó)塑料(2015年11期)2015-10-14 01:14:14
中國(guó)塑料(2015年9期)2015-10-14 01:12:17
中國(guó)塑料(2015年4期)2015-10-14 01:09:19
Coco薇(2015年1期)2015-08-13 02:47:34
