TPL2在免疫介導的炎性腸病中的研究進展*
2022-03-04 05:36:46董寧寧
醫學理論與實踐 2022年4期
董寧寧 陳 吉
1 內蒙古醫科大學,內蒙古包頭市 014010; 2 內蒙古包鋼醫院消化科
TPL2(Tumor progression locus 2)是一種癌基因的絲氨酸/蘇氨酸激酶。在結構上,TPL2 激酶由N端、激酶結構域、C端(羧基端)三部分組成。N端結構域可能負向調節TPL2穩定性,同時正向調節細胞轉化能力,主要靶向細胞內絲裂原激活蛋白激酶1/2(Mitogen-activation protein kinase,MAPK)(MEK1/2)的絲氨酸/蘇氨酸激酶結構域;羧基末端具有抑制激酶激活和負向調節TPL2穩定性和轉化能力的功能[1]。TPL2基因主要編碼Tpl-p58和Tpl-p52兩種蛋白質,分別由甲硫氨酸1 (M1)和甲硫氨酸30(M30)的替代翻譯產生[2]。TPL2可將各種細胞內和細胞外刺激傳遞給效應蛋白,進一步調節參與炎癥和免疫細胞募集、分化,同時激活促炎因子、趨化因子和生長因子的表達,調節炎癥和免疫細胞的功能[3]。盡管有大量關于TPL2在炎性,免疫性疾病中的研究,但TPL2在免疫介導的炎性腸病中的作用和潛在機制尚未得到充分的認識。本綜述旨在結合最新的文獻闡述TPL2在免疫介導的炎癥性腸病(Inflammtory bowel disease,IBD)中的作用以及潛在的機制。
1 TPL2與炎癥信號通路傳導
TPL2能夠被病毒、細菌感染、氧化應激、有害化學物質和促炎細胞因子等快速或瞬時激活[4]。外界刺激首先激活各種炎癥受體,包括腫瘤壞死因子受體 (Tumor necrosis factor receptor,TNFR)、CD40、白細胞介素1受體(Interleukin 1 receptor, IL1R)、模式識別受體(Pattern-recognition receptors, PRRs)[3]。隨后這些信號放大激活IκB激酶 (IκB kinase, IKK) 復合物,IKKα/IKKβ/IKKγ等[4]。IKKβ隨后使NF-κB1 p105磷酸化,進而導致TPL2激酶的釋放、磷酸化和激活[5]。TPL2 激活下游效應分子,包括細胞外信號調節激酶1和2 (ERK1/2)、p38α、JNK (c-Jun N-terminal kinase)和蛋白激酶B(Protein kinase B, PKBt),這些相關通路直接負責調控細胞因子、趨化因子、蛋白酶、生長因子和氧化應激轉錄因子等[6]。
促炎性細胞因子可以通過調節炎癥組織的細胞凋亡、改變血管內皮通透性、趨化炎癥細胞到炎癥組織,促使炎癥級聯放大。TPL2調控的相關基因包括細胞因子(TNF、IL-1α/β、IL-6、IL-17A、IFNγ和IL-10)、趨化因子[如IL-8、CXC-趨化因子配體1(CXCL1)、CCL2/MCP1和 CCL5/RANTES]、生長因子、環氧合酶 2和細胞周期蛋白D1和基質金屬蛋白酶 (MMP2、MMP3、MMP9和MMP13)[7]。TPL2調控的細胞因子和趨化因子進一步促進炎癥細胞的募集,這些細胞有助于炎癥微環境的形成[8]。TPL2調控的一些致癌途徑包括絲裂原活化蛋白激酶、活化B細胞的NF-κB (nuclear factor-kappa light-chain enhancer of activated B cells)、激活蛋白1(activator protein 1)、STAT3、雷帕霉素的機制靶標 (mechanistic target of rapamycin, mTOR)和CCAAT/增強子結合蛋白 β (C/EBPβ)[7]。因此,TPL2通過調節各種炎癥因子、轉錄因子和轉錄后機制,形成了一個正反饋回路,其可能對炎性腸道疾病的發生、發展起關鍵作用。見圖1。
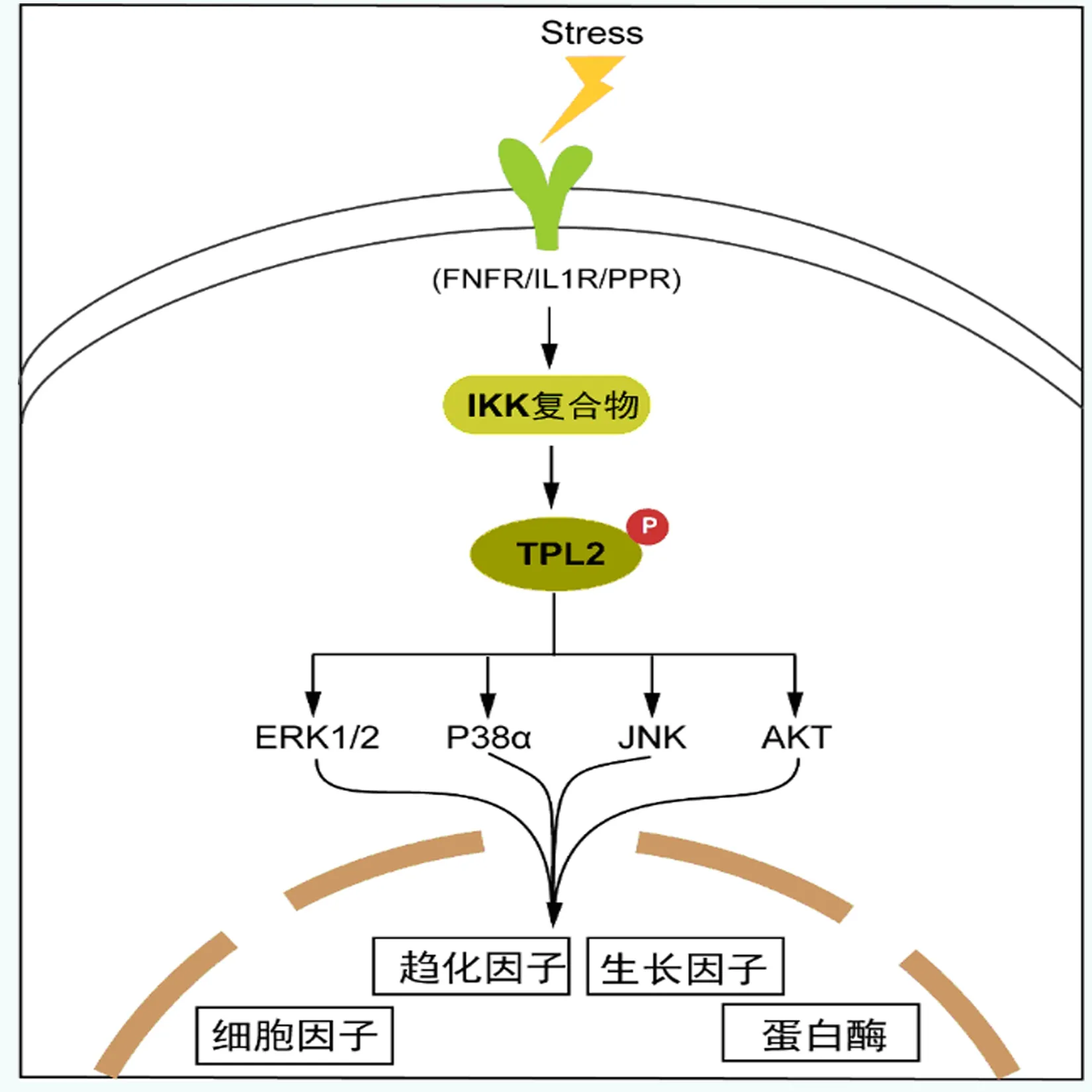
圖1 TPL2在炎癥信號通路中的作用
2 TPL2與免疫反應
TPL2最初被認為是一種致癌基因,隨后的研究顯示其可通過MEK-ERK MAPK信號通路來特異性激活和調節先天性和適應性免疫反應。絲裂原活化蛋白激酶 (Mitogen-activated protein kinase,MAP) 級聯反應是免疫激活和炎癥介質轉錄誘導的主要細胞內信號通路。腫瘤進展位點2 (TPL2/MAP3K8) 在MAP激酶中通過促進 ERK 依賴性誘導調節 IFN的 產生[9]。雖然TPL2 敲除動物表現出所有的免疫器官和免疫系統細胞亞群的比例都是正常的,同時他們對 T細胞依賴性和非依賴性抗原的抗體反應也是正常的[10]。但是TPL2是漿細胞樣樹突狀細胞產生 IFN-α和 CD4+T細胞分泌 IFNγ 所必需的,同時對巨噬細胞和 DC 中 IFN-β 的有效性調節反饋。據報道,TPL2缺陷型巨噬細胞可以通過轉錄和轉錄后調控來抑制TNF的產生[11],但這種作用具有細胞特異性,對DC細胞并沒有很好的效果。在巨噬細胞中,TPL2 通過與 NFκB1/p105的相互作用保持無活性形式,IκB激酶復合物的激活導致 TPL2 磷酸化及其從p105中分離。磷酸化的TPL2被釋放以激活 MEK-ERK信號通路。TPL2-/-巨噬細胞中LPS激活ERK的失敗導致細胞因子、趨化因子和其他參與先天性和適應性免疫調節的分子的表達缺陷[10]。此外,Watford 等人發現TPL2對CD4+T細胞產生IFN- γ的產生也極為重要[12]。近年研究顯示TPL2對CD8+T細胞的產生也至關重要[13]。除了干擾素外,TPL2還調節其他重要免疫介質的產生,如 TNF-α、IL-1β、IL-10、IL-12和 COX-2[6]。TPL2還可以誘導骨髓來源的樹突狀細胞P38 MAPK的產生[14]。因此,TPL2通過調節巨噬細胞,樹突狀細胞和NK細胞構建了第一道先天免疫反應。在自適應性免疫系統中,免疫系統通過CD40-TPL2-ERK信號通路介導B細胞產生免疫球蛋白,但其對LPS介導的B細胞凋亡的不敏感性,以及對IL-12介導的輔助T細胞分化作用較小。TPL2在免疫系統中的那些矛盾作用表明其免疫系統的調控具有明顯的細胞特異性以及兩面性的特點。
3 TPL2在免疫介導的炎性腸炎中的作用
IBD是一系列胃腸道的慢性炎癥性疾病,包括克羅恩病(Crohn’s disease,CD)、潰瘍性結腸炎(Ulcerative colitis,UC)和不確定性結腸炎。雖然IBD的原因尚不完全清楚,但目前其遺傳易感性已得到廣泛的認可,IBD 患者的一級親屬也被證明患 IBD 的風險增加了5倍[15]。除了遺傳易感性之外,先天性和適應性免疫機制的異常激活也被認為會導致 IBD的進展[16]。免疫介導疾病的一個標志是細胞因子分泌失調,而IBD表現為腸道免疫穩態和細胞因子分泌失調。T 細胞介導的免疫反應在 CD 和 UC 中均被放大。在 CD 中,炎癥是由放大的 T 輔助 (Th) 1 和 Th17 反應引發的;這會導致促炎細胞因子IL-17、IFN-γ 和TNF-α 的分泌,進而導致炎癥的自我循環及進展。在 UC 中,該反應是 Th2 介導的,導致 B 細胞和自然殺傷 T 細胞的更有效激活,并由 IL-5 和 IL-13 介導[15]。同時許多與 IBD 相關的基因位點也負責 T 細胞功能。
腸道屏障也與宿主先天免疫密切相關。這種環境由腸上皮細胞(腸上皮細胞、杯狀細胞、神經內分泌細胞、潘氏細胞和 M 細胞)和免疫細胞組成。在動物模型中,這種屏障的紊亂已被證明會導致IBD,例如在 Muc2缺陷小鼠中,它們無法從杯狀細胞分泌黏蛋白并發展為IBD[17]。潘氏細胞缺陷已被證明是通過NOD2基因異常介導的,與CD的發展有關。NOD2已被證明與細胞內病原體的自噬和清除有關,該途徑的破壞與 CD的發展密切相關[18]。黏膜屏障中的抗原呈遞細胞(APC)也與IBD的發展有關。CD患者已被證明具有減弱的巨噬細胞活性,這會損害中性粒細胞的活性并允許更多的細菌易位穿過腸道[15]。另一種類型的 APC(樹突狀細胞),已被證明會在IBD患者腸道的固有層中積聚。這種向淋巴濾泡的運輸受損阻止了適當的抗原采樣和免疫激活。
近年研究顯示TPL2激酶活性在其中起到重要作用。TPL2 介導先天免疫反應和 T 細胞反應,研究顯示TPL2 有助于小鼠中PRR 誘導的細胞因子分泌和抑制內毒素休克[12]。同時TPL2過表達會導致小鼠結腸炎。TPL2 活性受IKKβ 對 Thr290 的磷酸化調節;TPL2介導的MAPK 或 NF-κB 通路的激活可能取決于細胞類型、刺激和物種。在IBD中,TPL2 基因表達增加,將促進放大模式識別受體 (PRR) 介導的 ERK、JNK 和 NF-κB 信號通路的激活以及細胞因子的產生[19]。Watford等人研究顯示TPL2參與了 TGF-β誘導的 FoxP3 表達, TPL2抑制劑可以選擇性地靶向基于輔助性T細胞的炎癥[20]。TPL2激酶活性負責協調成纖維細胞對損傷的反應,并通過調節基底膜完整性來控制上皮組織穩態。
TPL2缺陷小鼠表現出腸道炎癥和對化學物質誘導的結腸炎的超敏反應。最近的研究顯示TPL2 缺陷小鼠炎癥增加可能是由于ABIN2-A20相互作用和信號傳導受損的結果。A20進一步誘導 IL-10表達和Treg細胞生成[21]。TPL2激酶是作為IL-10表達的正向調節因子,TPL2 活性喪失可能抑制 IL-10 介導的抗炎信號,導致腸道炎癥的增加,從而驅動腫瘤發生[22]。在TNF驅動的Crohn的炎性腸病疾病小鼠模型中,TPL2缺失將抑制的記憶CD4+和外周CD8+淋巴細胞的數量,并改善了疾病的發病和進展。雖然以前使用小鼠炎性疾病模型的研究表明TPL2主要是促炎功能,但在某些情況下,TPL2抑制促炎細胞因子和功能[22]。因此,TPL2對免疫介導的相關炎性腸病具有兩面性的作用效果,作為促炎介質,TPL2 響應 LPS 正向調節 TNF-α 的表達和轉運,而對內毒性休克具有一定的抑制效果。
4 TPL2靶向藥物的開發
由于TPL2在免疫系統的重要作用,因此其靶向藥物的開發可能是許多類型炎癥腸病的有效治療策略。TPL2與其他激酶的同源性較低,不受非特異性激酶抑制劑(星形孢菌素)的抑制,并且是唯一已知的在其ATP結合區具有脯氨酸而不是甘氨酸的人類激酶,這些都使其成為具有選擇性的藥物靶點。ATP競爭性TPL2特異性抑制劑,如1,7-萘啶-3-碳腈、噻吩吡啶等是基于免疫治療為基礎,表現出良好的特異性抗炎效果[23]。通常情況下,蛋白激酶的催化域可以將磷酰基從三磷酸腺苷(ATP)轉移到底物上,ATP與二價金屬離子如Mn2+、Mg2+、Ca2+、Fe2+、Co2+和 Ba2+的結合是催化磷酸轉移過程的重要步驟。大多數激酶與Mg2+的親和力高,而TPL2多將Mn2+作為 ATP 金屬輔助因子,表明Mn2+ATP與TPL2 ATP 具有高親和力的結合位點。TPL2 激酶具有明顯的結構特征,這有利于開發高度特異性、有效和選擇性的TPL2激酶抑制劑。例如,TPL2激酶與其他激酶的同源性相對較低,并且在激酶結構域上含有脯氨酸而不是甘氨酸。當與各種激酶抑制劑結合時,該激酶還顯示出具有更靈活的活性結構位點和獨特的激酶結構域折疊[24]。
TPL2作為連接炎癥和免疫的關鍵分子。盡管TPL2在炎性相關疾病的發生取得了巨大進展,但目前在臨床前和臨床試驗中還沒有TPL2拮抗劑。主要是存在以下幾個方面的因素阻礙了現有TPL2激酶抑制劑的臨床轉化:(1)尚未明確TPL2結構信息和調控TPL2復合物形成和折疊的分子機制;(2) 缺乏預測和監測患者對TPL2激酶抑制劑選擇性反應的分子生物標志物;(3)TPL2抑制劑的成本。因此,未來研究應重點突破TPL2復合物形成和折疊的分子機制,通過生物技術、分子技術以及人工智能技術進一步解析TPL2復合體的3D結構。此外,基于結構的藥物設計和配體的藥物設計方法來優化和開發更有效的TPL2激酶抑制劑,同時建立科學的TPL2體外生成和純化的技術路線與方法。
總而言之,TPL2作為炎癥及免疫相關信號通路的樞紐,為靶向控制炎癥信號通路提供了潛在的治療策略。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
學苑創造·A版(2020年9期)2020-10-13 09:41:02
中國生殖健康(2019年3期)2019-02-01 06:12:26
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
鑿巖機械氣動工具(2016年3期)2016-03-01 04:00:25
海軍航空大學學報(2015年3期)2015-11-11 17:20:00
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
七彩語文·畫刊(2012年3期)2012-04-29 00:00:00
