全二維氣相色譜-飛行時間質(zhì)譜法測定汽油中21種抗爆添加劑
2022-03-04 05:08:40劉一龍薛秋紅管曉倩王凱歌馮真真周龍龍王群威
分析測試學報 2022年2期
關鍵詞:標準
劉一龍,薛秋紅*,管曉倩,王凱歌,馮真真,周龍龍,王群威
(1.青島海關技術中心,山東 青島 266500;2.寧波海關技術中心,浙江 寧波 315048)
隨著汽車工業(yè)的不斷發(fā)展,發(fā)動機效率的不斷提高和減少CO2排放的環(huán)境要求導致對汽油辛烷值的要求越來越高[1],向汽油中添加抗爆添加劑是提升汽油辛烷值最經(jīng)濟有效的方法[2-3]。但各種抗爆添加劑在改善汽油辛烷值的同時,存在會引起環(huán)境污染、損害汽車發(fā)動機等各種問題[4],因此我國車用汽油產(chǎn)品標準GB 17930-2016[5]對抗爆添加劑的種類和含量做出了嚴格限制。
目前,針對汽油中非金屬抗爆添加劑的檢測方法主要有紅外光譜法[6-7]、選擇性檢測器氣相色譜法[8-10]、閥切換反吹氣相色譜法[11-13]、二維中心切割氣相色譜法[14-16]、氣相色譜-質(zhì)譜聯(lián)用法[17-18]等方法。這些分析方法中,紅外光譜法具有分析速度快、操作簡單、重復性好、分析成本低、不破壞樣品、容易實現(xiàn)在線分析等特點[19],在快速分析中具有巨大的應用潛力。選擇性檢測器氣相色譜法只對特定化合物有響應,在單一類別化合物分析中具有更好的選擇性和靈敏度。閥切換反吹氣相色譜法和二維中心切割氣相色譜法是目前采用較多的分析方法,前者還是我國車用汽油產(chǎn)品標準中指定的含氧化合物檢測仲裁方法(NB/SH/T 0663-2014[20])。這兩種二維氣相色譜方法分離能力較一維色譜有所提升,可實現(xiàn)多種特定目標化合物的二次分離。氣相色譜-質(zhì)譜聯(lián)用法可以利用質(zhì)譜的定性能力和選擇性提升方法的鑒別性能和檢測靈敏度。
全二維氣相色譜飛行時間質(zhì)譜技術具有高靈敏度、高分離能力、高峰容量等特點,在眾多領域得到了廣泛應用,在石油化工領域主要用于油品中烴類族組成分布檢測[21-24],在汽油中抗爆添加劑檢測方面的研究較少[25]。
本研究應用全二維氣相色譜-飛行時間質(zhì)譜法檢測汽油中的非金屬抗爆添加劑類化合物,基于全二維色譜柱容量大和分離能力強的特點,實現(xiàn)了汽油中21 種多類別(醇類、醚類、酯類、胺類、酮類等)抗爆添加劑的同時檢測。該方法所檢測的化合物種類與濃度范圍可滿足相關國家標準對汽油中禁用和限用抗爆添加劑的檢測要求。
1 實驗部分
1.1 儀器與試劑
全二維氣相色譜-飛行時間質(zhì)譜儀(GC×GC-TOF MS),配置MPS多功能自動進樣器(德國Gerstel公司),7890A 氣相色譜儀(美國Agilent 公司),雙階四噴熱調(diào)制器、二維爐溫箱、Pegasus 4D TOF MS、ChromaTOF?V4.72.0.0 工作站軟件(美國LECO 公司);BSA224S 電子天平(精度0.1 mg,德國Sartorius公司);100、1 000、5 000 μL 移液器(德國Eppendorf 公司);DB-5MS 色譜柱(美國Agilent 公司),BPX-50 色譜柱(澳大利亞SGE 公司),一、二維柱接頭:SilTite μ?union 0.4 to 0.4(澳大利亞Trajan公司)。
甲醇(MeOH)、乙醇(EtOH)、正丙醇(NPA)、異丙醇(IPA)、正丁醇(NBA)、異丁醇(IBA)、叔丁醇(TBA)、仲丁醇(SBA)、仲戊醇(SAA)、叔戊醇(TAA)、甲基叔丁基醚(MTBE)、乙基叔丁基醚(ETBE)、甲基叔戊基醚(TAME)、乙基叔戊基醚(TAEE)、二異丙基醚(DIPE)、丙酮(DMK)、碳酸二甲酯(DMC)、3-甲氧基異丁酸甲酯(Methyl 3-methoxyisobutyrate)、乙酸異丁酯(IBAC)、N-甲基苯胺(NMethylaniline)、N,N-二甲基苯胺(N,N-Dimethylaniline)、乙二醇二甲醚(DME)、異辛烷、二甲苯(中國西亞試劑公司,色譜純)。汽油樣品為市場銷售的汽油產(chǎn)品。
1.2 實驗條件
1.2.1 色譜條件一維柱:DB-5MS色譜柱(30 m×0.25 mm×0.25 μm);二維柱:BPX-50色譜柱(1 m × 0.1 mm × 0.1 μm)。進樣量:0.2 μL;載氣:氦氣(純度≥99.999%);進樣方式:分流進樣;分流比:150∶1;載氣流速:1.0 mL/min;進樣口溫度:200 ℃;柱溫:初始溫度35 ℃保持6 min,以5 ℃/min 升至50 ℃,保持4 min,然后以30 ℃/min 升至200 ℃,保持5 min;二維柱補償溫度:+5 ℃;調(diào)制器補償溫度:+15 ℃;調(diào)制周期:6 s;熱吹時間:1.5 s;傳輸線溫度:280 ℃。
1.2.2 質(zhì)譜條件離子源:電子轟擊(EI)源;離子掃描范圍:30~250 amu;采集頻率:100 Hz;檢測器電壓:1 700 V;電離能:70 eV;離子源溫度:230 ℃。
1.3 實驗方法
1.3.1 標準溶液的配制為模擬真實汽油狀態(tài),將所有標準化合物分為兩組,其中A 組包括14 種常規(guī)醚類和醇類抗爆添加劑(MeOH、EtOH、NPA、IPA、NBA、IBA、TBA、SBA、SAA、TAA、MTBE、ETBE、TAME、DIPE)以及內(nèi)標化合物(DME);B 組包括7 種非常規(guī)抗爆添加劑(DMK、TAEE、DMC、3-甲氧基異丁酸甲酯、IBAC、N-甲基苯胺、N,N-二甲基苯胺)和內(nèi)標化合物(DME)。用異辛烷-二甲苯混合溶劑(體積比63∶37)模擬汽油基體,重量法配制質(zhì)量分數(shù)分別為0.1%、0.5%、1%、2%、5%、12%、20%的A組和B組系列標準儲備溶液。
分別移取A 組和B 組系列標準儲備溶液各5 mL 于100 mL 容量瓶中,以異辛烷為溶劑定容至刻度,得到系列混合(A 組和B 組)標準使用液S1 ~ S7,其質(zhì)量濃度水平約為50、250、500、1 000、2 500、6 000、10 000 mg/L。混合標準使用液配制情況詳見表1。
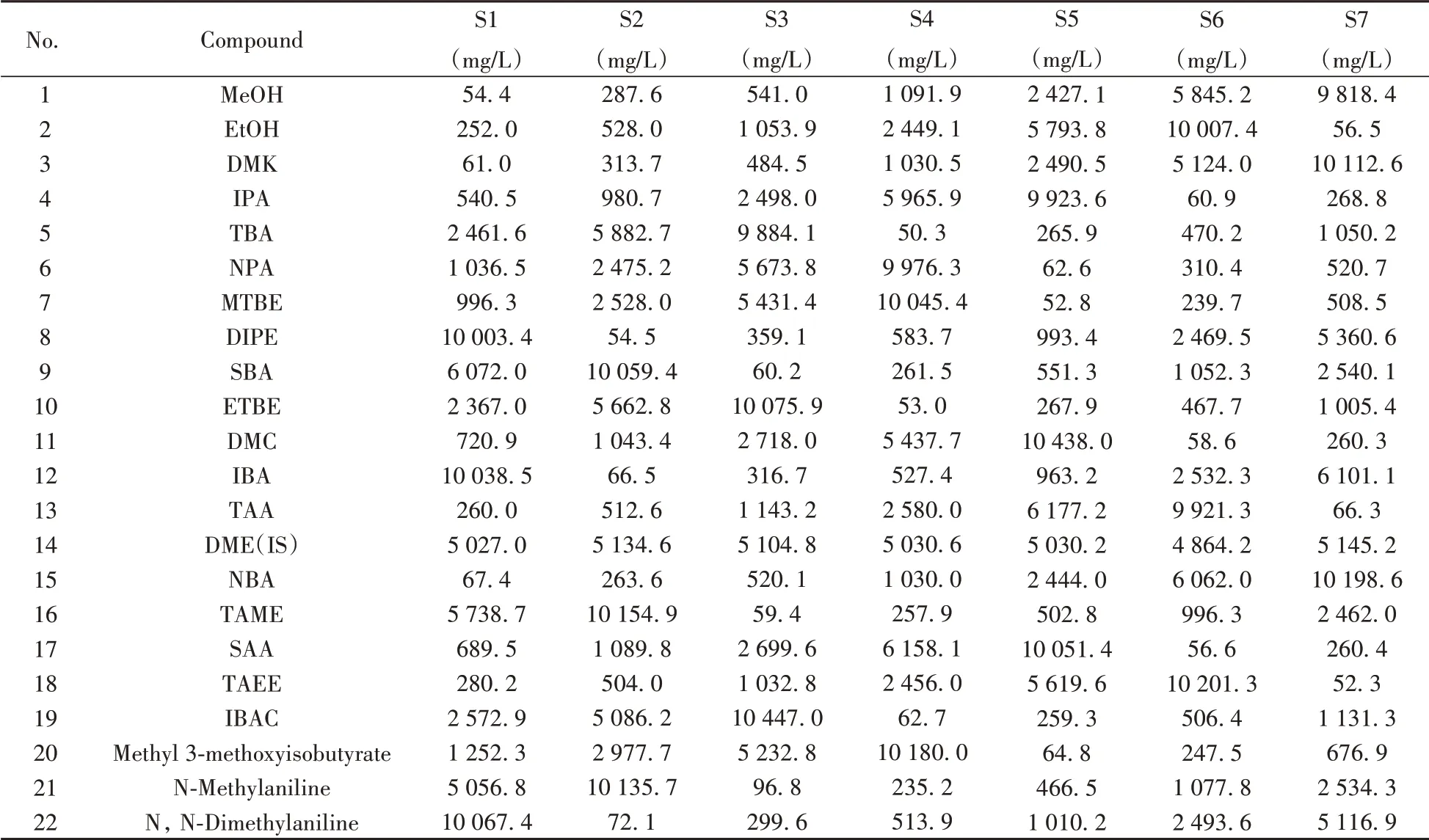
表1 抗爆添加劑混合標準使用溶液S1~S7的質(zhì)量濃度Table 1 Concentrations of antiknock additive standard solutions S1-S7
1.3.2 樣品前處理考慮到汽油樣品的揮發(fā)性,通過重量差減的方式,準確稱取約5 g 樣品和0.25 g內(nèi)標化合物(DME)至100 mL容量瓶中,用異辛烷定容至刻度,混勻后上機分析。
2 結(jié)果與討論
2.1 一維色譜柱升溫程序優(yōu)化
目標抗爆添加劑種類涵蓋醚類、醇類、酯類、胺類、酮類等多種化合物,其極性差異較大,丙酮和MTBE 的沸點最低,約為56 ℃,N-甲基苯胺和N,N-二甲基苯胺的沸點最高,約為195 ℃左右,大部分目標化合物沸點集中在60~120 ℃范圍內(nèi),在升溫程序前半段出峰較集中。對一維色譜柱升溫程序進行優(yōu)化,考察了初始溫度為35、40、50、60 ℃和初始溫度保持時間分別為2、4、6 min 時的色譜峰分離情況。結(jié)果顯示,隨著一維柱初始溫度的增加和初始溫度保持時間的縮短,低沸點化合物的分離逐漸變差,22 種目標化合物中有18 種在一維柱溫度達到50 ℃前流出,當初始溫度為35 ℃且保持時間為6 min時低沸點化合物的分離效果最好,當初始溫度在50 ℃以上或初始溫度保持時間為4 min以下時,無論另一條件如何改變,在一維柱上都會出現(xiàn)較明顯的共流出現(xiàn)象。最終選擇初始溫度35 ℃保持6 min,以5 ℃/min 升至50 ℃,保持4 min,然后以30 ℃/min 升至200 ℃,保持5 min 作為一維柱升溫程序。
使用優(yōu)化后的色譜條件(見“1.2”)分析標準樣品,22 種目標化合物的總離子流色譜圖如圖1所示。
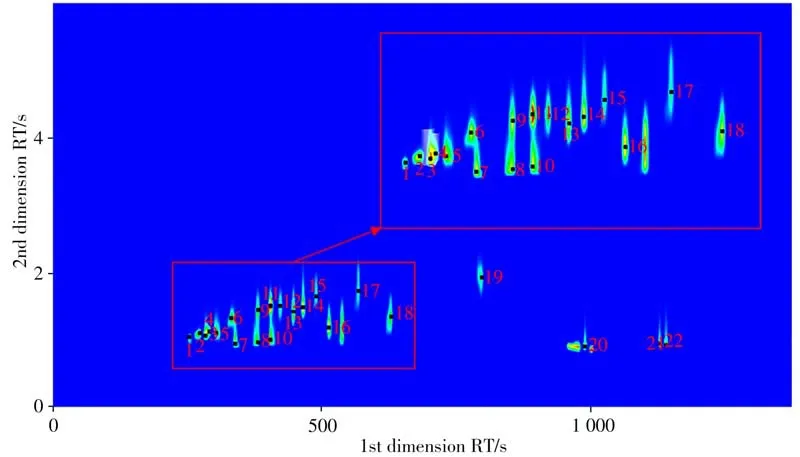
圖1 22種目標化合物的總離子流(TIC)色譜圖Fig.1 TIC chromatogram of 22 analytes
2.2 全二維分析模式與一維分析模式的比較
使用GC×GC-TOF MS的一維分析模式和全二維分析模式在“1.2”條件下分析同一標準溶液,結(jié)果顯示,一維模式下,DMK(保留時間285.35 s)和IPA(保留時間287.94 s)、DIPE(保留時間379.81 s)和SBA(保留時間382.77 s)、DMC(保留時間405.74 s)和ETBE(保留時間408.77 s)會出現(xiàn)共流出現(xiàn)象,其他目標化合物峰也與汽油基質(zhì)組分峰存在不同程度的共流出情況;但全二維分析模式下,大部分目標化合物有較好的分離效果,可與汽油組分基質(zhì)分離。對于少量分離效果較差的化合物,如DMK 和IPA,可以通過特征離子的選擇實現(xiàn)有效識別和定量,如DMK 使用58 amu、IPA 使用45 amu 作為特征離子時不存在相互干擾。
使用GC×GC-TOF MS 全二維模式分析,對于僅依靠色譜難以分離、主要特征離子又存在相互干擾的化合物,可以選擇相對豐度更低但特異性更強的特征離子進行定量。如DIPE和SBA雖然能得到分離但保留時間較為接近,可能發(fā)生相互干擾,對于SBA,其相對豐度在10%以上的離子均會受到DIPE離子碎片的干擾,這種情況下使用傳統(tǒng)一維GC-MS 很難選擇合適的特征離子進行定量分析。但得益于GC×GC-TOF MS 的高靈敏度,化合物的每個離子碎片都有較高的絕對響應值,即使其特征離子相對豐度較低,仍然具有足夠的信噪比進行定量計算,故可以選擇相對豐度較低但不會受DIPE 干擾的55 amu離子作為SBA的特征離子進行定量。最終選擇的21種抗爆添加劑和內(nèi)標化合物的定量離子如表2所示。
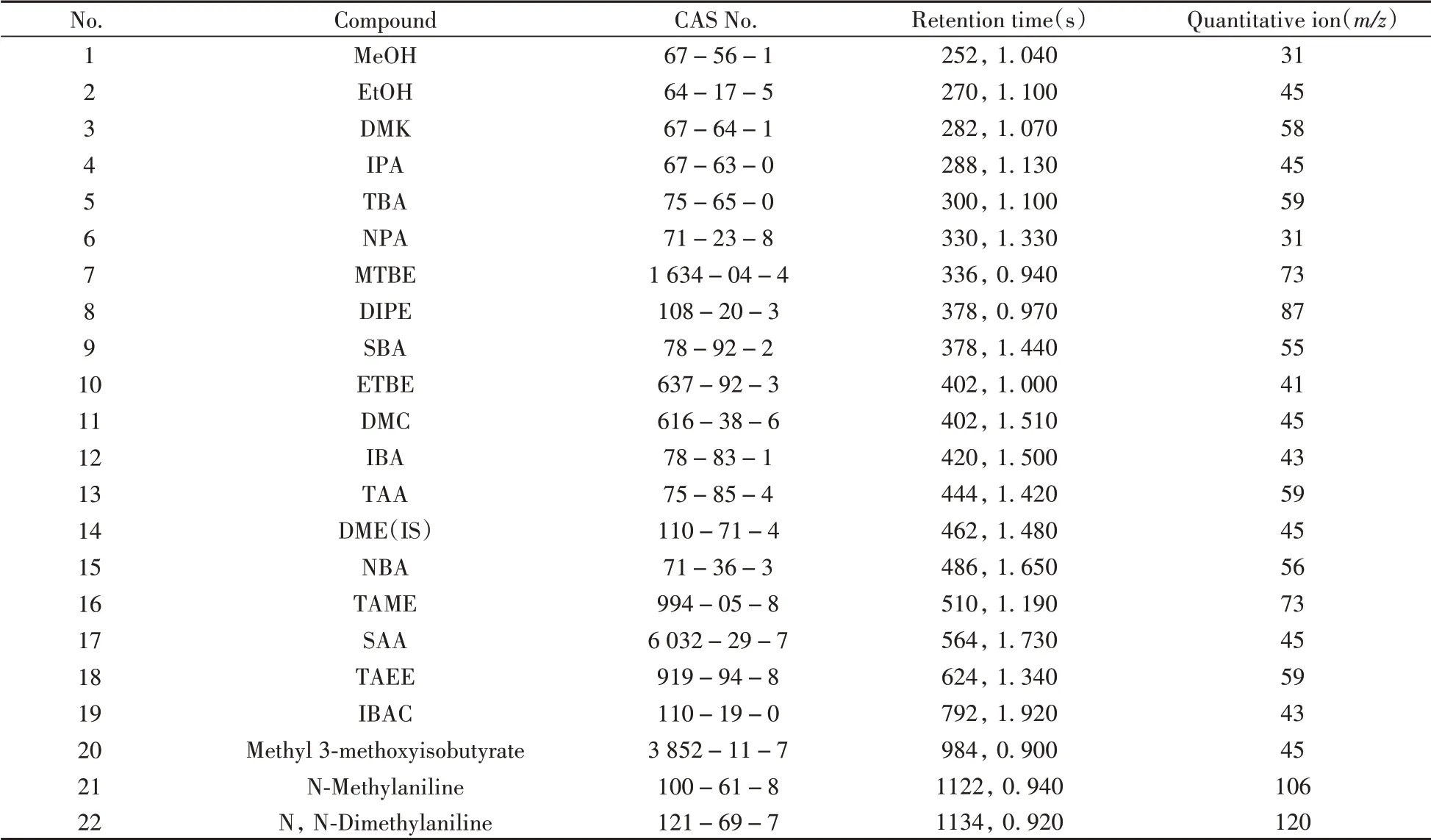
表2 21種抗爆添加劑和內(nèi)標化合物(IS,DME)的保留時間、定量離子Table 2 Retention times and quantitative ions of 21 antiknock additives and internal standard(IS,DME)
2.3 定量方式選擇
考察了對標準溶液使用內(nèi)標法峰面積定量、外標法峰面積定量、內(nèi)標法峰高定量、外標法峰高定量4 種方式進行定量的差異。使用內(nèi)標法定量的優(yōu)點是可以忽略進樣量和儀器漂移的微小變化,而使用外標法定量的前處理步驟較簡單,不需要再單獨添加內(nèi)標化合物;使用峰面積定量不易受峰型變化的影響,使用峰高定量則可在全二維模式下忽略化合物被調(diào)制至多個周期時的調(diào)制峰合并情況,不需要再進行繁瑣的確認。
分別使用上述4種定量方式,以普通最小二乘法計算標準曲線的線性相關系數(shù)(r2)并將其按大小排序。結(jié)果顯示,4種定量方式標準曲線r2的順序為:內(nèi)標法峰面積>外標法峰面積>內(nèi)標法峰高>外標法峰高。對于21 種抗爆添加劑,內(nèi)標法峰面積定量的r2均大于0.996,外標法峰面積定量的r2均大于0.992,內(nèi)標法峰高定量的r2均大于0.96,外標法峰高定量的r2均大于0.95。綜上,內(nèi)標法的r2總體優(yōu)于外標法,峰面積定量的r2總體優(yōu)于峰高定量。這說明儀器漂移和峰形變化對定量結(jié)果的影響真實存在,雖然采用外標法和峰高定量會減少工作量,但使用內(nèi)標法和峰面積定量可以有效改善標準曲線的線性。
2.4 標準曲線擬合方式選擇
在線性回歸模型中,使用普通最小二乘法擬合計算時需符合高斯-馬爾柯夫假定[26],即測量誤差滿足零均值、同方差且互不相關的條件。而在分析化學實驗中,誤差項的方差往往隨著自變量濃度的增加而增加[27],即存在異方差性,這時使用普通最小二乘法擬合會導致低濃度樣品計算結(jié)果的相對偏差較大,模型的預測結(jié)果不符合實際情況,而采用加權(quán)最小二乘法可以有效改善這種情況[28]。
馬爾柯夫估計是加權(quán)最小二乘法的無偏有效估計,在一元線性回歸模型中,其權(quán)重值與測量值誤差的方差成反比,測量值誤差的方差可以通過多次重復測量或者迭代計算得到。但在分析化學實際實驗中,評估測量值誤差的方差時過程較為繁瑣,一般取濃度值x或測量值y的倒數(shù)1/xi或1/yi、其平方的倒數(shù)作為權(quán)重值[27]。本文考察了使用普通最小二乘法和以1/xi、1/xi2為權(quán)重的加權(quán)最小二乘法對標準曲線進行擬合時對計算結(jié)果的影響。以TBA 為例,分別使用這兩種方法擬合標準曲線一元線性方程,對每個標準點濃度進行計算,結(jié)果如表3所示。
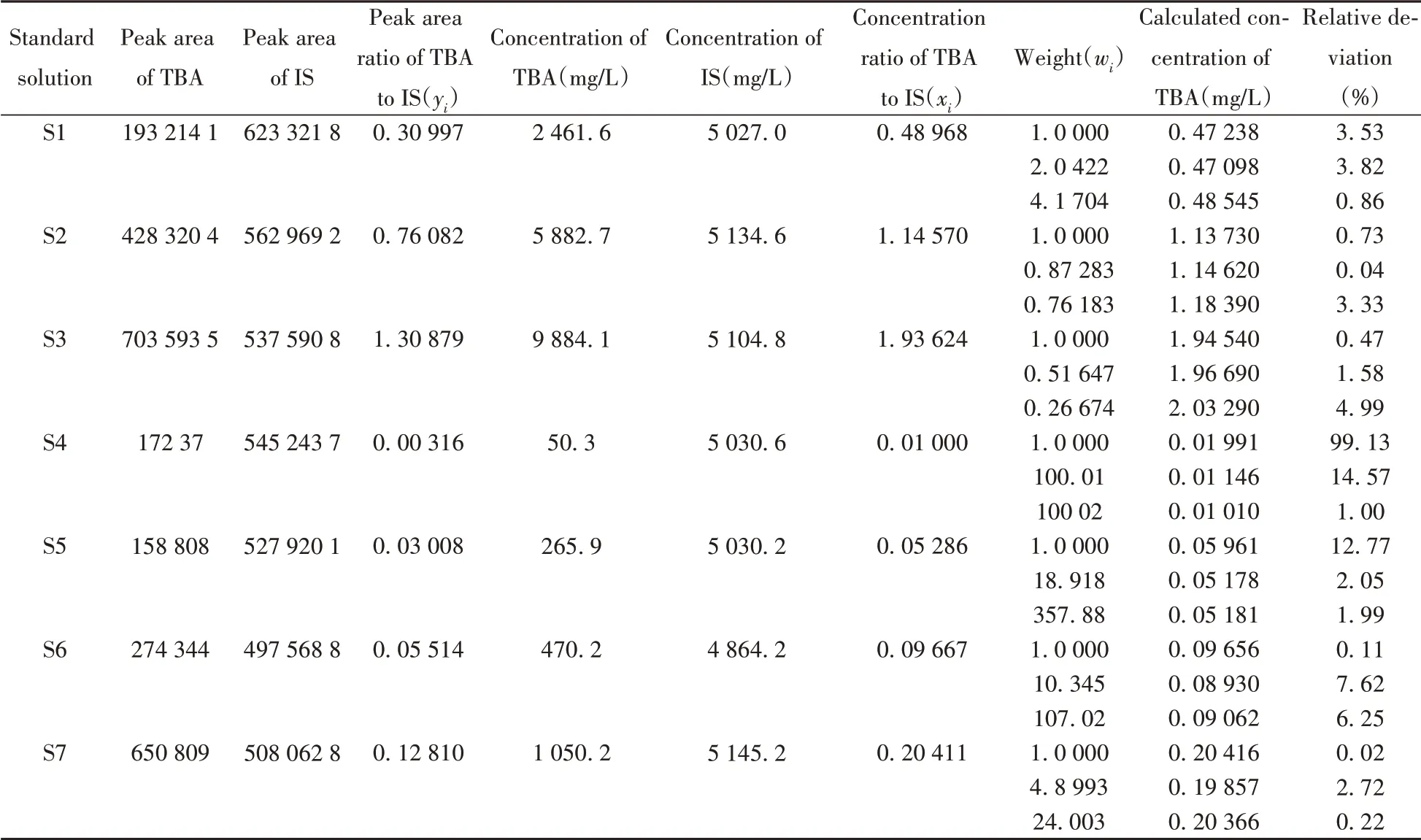
表3 不同權(quán)重下標準曲線對TBA計算結(jié)果的相對偏差Table 3 Relative deviations of TBA concentration calculated by standard curves with different weights
研究發(fā)現(xiàn),使用普通最小二乘法(即權(quán)重全部為1)和以1/xi、1/xi2為權(quán)重的加權(quán)最小二乘法得到的標準曲線線性系數(shù)(r2)分別為0.999 90、0.999 70、0.999 22;使用3種標準曲線計算TBA 質(zhì)量濃度時,低濃度標準溶液S4、S5的計算結(jié)果與通過重量法配制得到的濃度理論值的相對偏差依次減小,同時高濃度標準溶液中對應相對偏差值有不同程度增加,但仍小于10%。可以看出,使用加權(quán)最小二乘法擬合標準曲線可以有效改善低濃度溶液計算結(jié)果的準確性。
對7個標準溶液中21種抗爆添加劑的共147個濃度值分別使用普通最小二乘法和以1/xi、1/xi2為權(quán)重的加權(quán)最小二乘法擬合標準曲線,計算其濃度值和相對偏差,結(jié)果如表4所示。結(jié)果表明,使用加權(quán)最小二乘法計算標準溶液濃度的結(jié)果顯著優(yōu)于普通最小二乘法,且以1/xi2為權(quán)重的計算結(jié)果優(yōu)于以1/xi為權(quán)重的計算結(jié)果。

表4 不同權(quán)重下標準溶液計算結(jié)果的相對偏差分布Table 4 Relative deviation distribution of standard solutionscalculated by standard curves with different weights
2.5 線性范圍、檢出限、定量下限
按照優(yōu)化后的儀器條件使用GC×GC-TOF MS全二維分析模式分析抗爆添加劑標準使用液S1~S7,以DME為內(nèi)標,抗爆添加劑峰面積和內(nèi)標化合物峰面積的比(y)為縱坐標,抗爆添加劑濃度和內(nèi)標化合物濃度的比(x)為橫坐標,以1/xi2為權(quán)重使用加權(quán)最小二乘法擬合得到標準曲線。結(jié)果顯示,21種抗爆添加劑在約50~10 000 mg/L質(zhì)量濃度范圍內(nèi)線性良好,r2均大于0.995。按照“1.3.2”樣品前處理方法計算,方法對樣品中抗爆添加劑的含量檢測范圍約為0.1% ~ 20.0%。分別以3 倍(S/N≥3)和10 倍(S/N≥10)信噪比計算方法的檢出限(LOD)和定量下限(LOQ),得到方法的LOD為0.000 4%~0.014 1%,LOQ為0.001 4%~0.046 9%。
2.6 加標回收率及相對標準偏差
按照“1.3.1”方法向同一個不含待測化合物的汽油樣品中分別添加質(zhì)量分數(shù)約為0.5%、2.0%、5.0%、10.0%的A 組14 種和B 組7 種抗爆添加劑,采用交叉配制的方法使樣品中化合物添加總量大致相等,最終配制成4 個水平的樣品加標溶液,按照“1.3.2”方法進行前處理,使用“1.2”儀器條件進行測定,每個加標樣品重復測定6次,其回收率數(shù)據(jù)見表5。結(jié)果表明,21種抗爆添加劑的加標回收率為91.1%~109%,相對標準偏差(RSDs,n=6)為0.80%~4.9%。
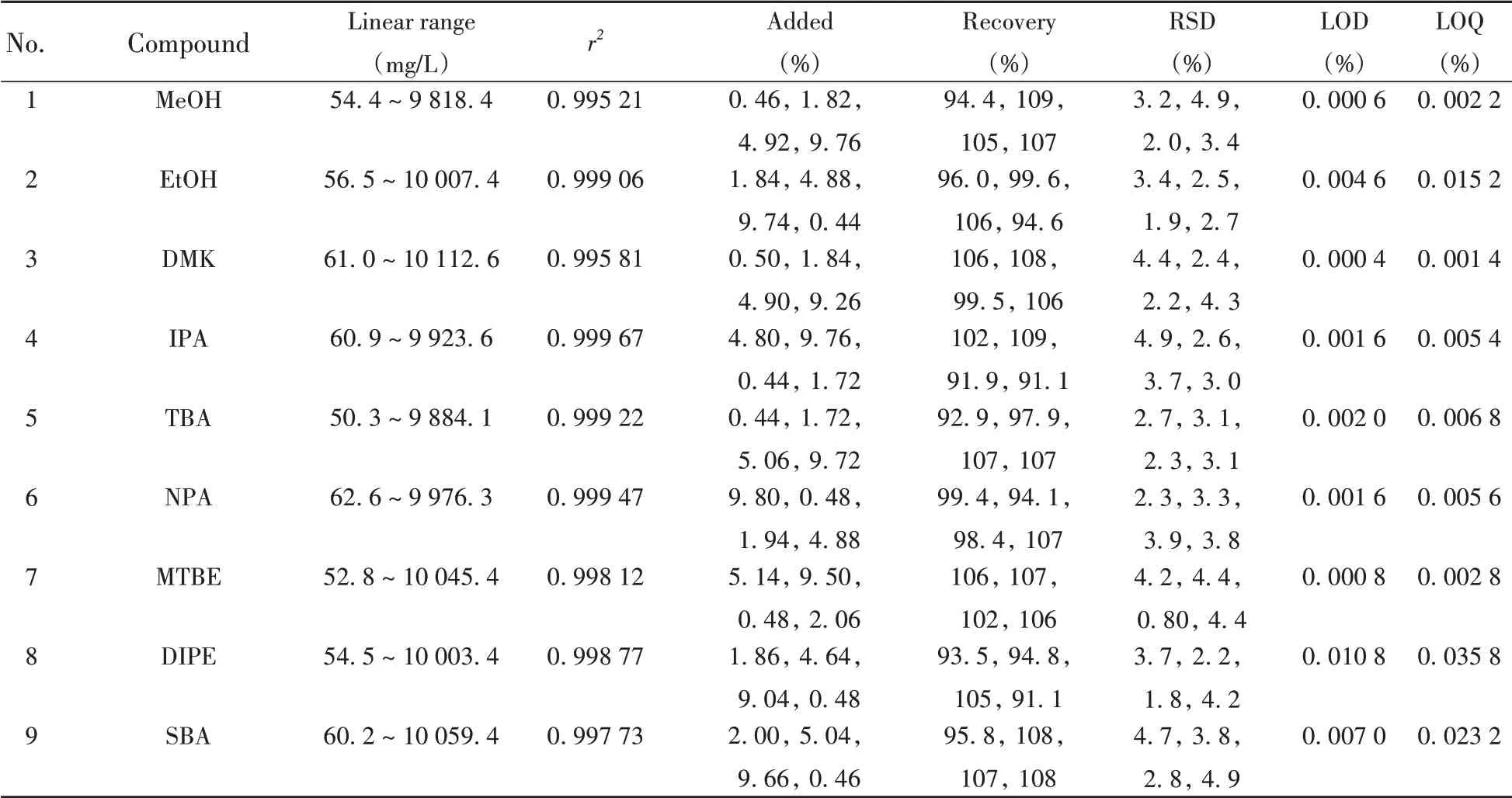
表5 21種抗爆添加劑的線性范圍、線性系數(shù)、加標回收率、相對標準偏差(RSDs)、檢出限(LODs)及定量下限(LOQs)Table 5 Linear ranges,correlation coefficients,recoveries,RSDs,LODs and LOQs of 21 antiknock additives
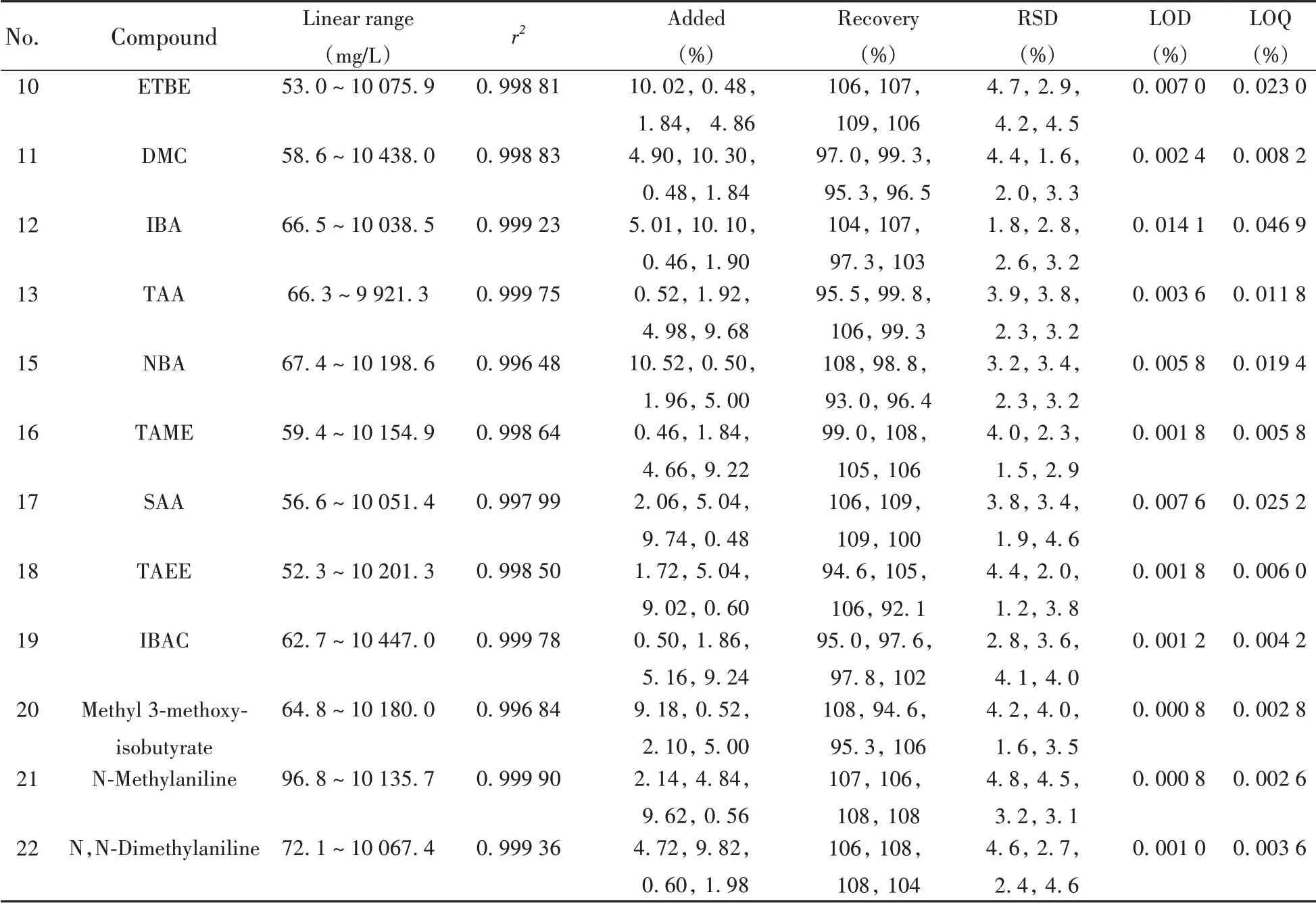
(續(xù)表5)
2.7 實際樣品檢測
使用本文建立的方法對加油站實際購買的92#、95#和98#汽油進行分析,結(jié)果在3種汽油產(chǎn)品中均檢出MTBE,其含量分別為3.14%、8.64%、12.48%。
3 結(jié) 論
本文建立了汽油中21種抗爆添加劑的全二維氣相色譜-飛行時間質(zhì)譜檢測方法,與傳統(tǒng)分類別單獨檢測方法相比,本法不僅檢測的化合物種類和數(shù)量更多,而且方法更加靈敏、可靠,極大提升了抗基體干擾能力。一次進樣即可實現(xiàn)GB 17930-2016《車用汽油》中規(guī)定的多種限用化合物的同時檢測,可以滿足市場產(chǎn)品質(zhì)量抽查和產(chǎn)品技術研發(fā)的檢測需要。此外,利用加權(quán)最小二乘法擬合標準曲線,比較了不同權(quán)重因子下結(jié)果準確度的差異,改善了對低濃度結(jié)果的定量準確性。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
當代陜西(2019年8期)2019-05-09 02:22:48
上海建材(2019年1期)2019-04-25 06:30:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
專用汽車(2016年4期)2016-03-01 04:13:43
質(zhì)量與標準化(2015年9期)2015-12-31 11:41:40
中國質(zhì)量與標準導報(2014年4期)2014-03-11 19:54:25
中國質(zhì)量與標準導報(2014年10期)2014-02-28 22:25:47
中國質(zhì)量與標準導報(2014年7期)2014-02-28 22:24:39
