PVA-g-mPEG接枝聚合物的制備及其防藥物濫用性能
2022-03-09 00:41:24楊璐李效文李鳳和姚日生
化工進展 2022年2期
楊璐,李效文,,李鳳和,姚日生
(1 合肥工業大學食品與生物工程學院,安徽 合肥 230009;2 安徽安生生物化工科技有限責任公司,安徽 合肥 230002)
由于阿片類藥物的濫用問題日益嚴重,防濫用制劑(abuse deterrent formulations,ADFs)應運而生。ADFs 主要是通過一系列的改造降低阿片類藥物濫用的可取性,具體手段包括添加物理屏障使得藥片很難被研磨粉碎、設置化學屏障通過形成黏性凝膠降低藥物溶劑提取率、添加拮抗劑或厭惡劑、制成前藥等從而消除藥物的欣快感。目前市場上批準的ADFs 產品主要是使用超高分子量聚環氧乙烷(PEO)添加物理化學屏障來阻止藥物濫用。PEO是一種非離子型、無毒性的親水性聚合物,分子量在10萬~700萬之間。當它與水接觸時,迅速水化形成黏性凝膠,阻止注射濫用。PEO的熔點在65~70℃之間,玻璃化轉變溫度在-50~-57℃之間,因此在機械應力作用下發生塑性變形而不是脆性斷裂,從而可以避免藥片因施加外力發生粉碎。
但超高分子量PEO 的制備條件較為苛刻,催化體系復雜昂貴或危險性高,工藝實現難度大。目前,僅有美國陶氏化學公司等歐美日企業能實現大規模工業生產,因此迫切需要開發新的防濫用材料。通過分子設計可得到具有特殊分子結構和不同優異性能的接枝型聚合物一直是高分子研究的一個熱點。如德國化學公司巴斯夫旗下的產品Kollicoat IR是采用自由基聚合先將聚乙酸乙烯酯鏈通過碳碳鍵接枝到PEG 骨架上,再水解形成PVA 側鏈得到的PVA--PEG 接枝共聚物,因其具有良好的成膜性而廣泛應用于普通包衣成膜劑。
鑒于防濫用制劑的所需特性,本文選用了親水性極強、但幾乎不溶于有機溶劑的具有良好生物相容性的聚乙烯醇(PVA)作為接枝主鏈,通過偶聯接枝法合成了聚乙烯醇接枝單甲氧基聚乙二醇(mPEG)聚合物(PVA--mPEG),并以高水溶性藥物鹽酸二甲雙胍作為模型藥物,考察了防濫用效果,開發了新的防濫用輔料產品,應用前景廣闊。
1 材料和方法
1.1 材料和儀器
單甲氧基聚乙二醇(mPEG,=4000)、聚乙烯 醇(PVA,=84000~89000, 醇 解 度86%~89%)、三氟化硼乙醚溶液(BF-EtO,48%BF,52%EtO)、環氧氯丙烷(ECH,99%),Aladdin 試劑有限公司;鹽酸二甲雙胍(>99.9%),廣州柏森藥業有限公司;微晶纖維素、聚乙烯吡咯烷酮(PVP k30)、硬脂酸鎂和二氧化硅,Adamas-beta試劑有限公司;其他試劑均為分析純或色譜純。
Nicolet-6700 型傅里葉紅外光譜儀(FTIR),美國ThermoNicolet 公司;VNMRS-600 型超導核磁共振波譜儀,美國安捷倫科技公司;DSC-Q2000差熱示差掃描量熱儀,美國TA 公司;PANalytical X-Pert PRO MPD 型固定靶X 射線衍射儀,荷蘭帕納科公司;ZPJ-4片劑智能四用儀,天津恒創立達公司;LABSOL-DB-CHS高效液相色譜儀,日本島津公司。
1.2 PVA-g-mPEG的制備
將真空干燥后的50g mPEG 倒入裝有機械攪拌器、溫度計和回流冷凝管的四口燒瓶中。在50℃下,向燒瓶中加入0.4mL BF-EtO 以及一定量的ECH,升溫至60℃反應5h。然后通過旋蒸去除過量的ECH,即得到mPEG 的表氯醇化中間體(mPEG-Cl)。通過滴定法測定mPEG-Cl 的氯原子含量間接測定mPEG的羥基轉化率。
在攪拌下,向四口燒瓶中依次加入80g/L PVA水溶液50g 和5g mPEG-Cl,升溫至反應溫度后,加入100g/L NaOH 水溶液1.5mL,保溫反應10h。反應結束后,先用鹽酸調節溶液至中性,再用乙醇索氏提取未反應的側鏈mPEG-Cl,得到PVA--mPEG 接枝聚合物粗產物。隨后產物使用1000kDa 透析袋透析60h,50℃真空干燥后得到了純化的PVA--mPEG接枝聚合物。合成路線如圖1所示,根據式(1)采用增重法計算接枝率()。
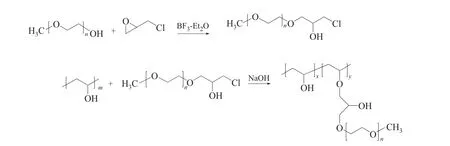
圖1 PVA-g-mPEG接枝聚合物的合成原理
1.3 PVA-g-mPEG負載藥物
以鹽酸二甲雙胍作為模型藥物,將PVA-mPEG與藥物按質量比3∶2溶解于一定量的二甲基亞砜中,將上述均一溶液緩慢滴加到冰丙酮中,抽濾(保留濾液,待測其中鹽酸二甲雙胍含量),50℃真空干燥,得到負載藥物的PVA--mPEG 絮狀物。
根據標準曲線采用高效液相色譜測定濾液中鹽酸二甲雙胍的含量:C18 柱(5μm,規格4.6mm×250mm),檢測波長233nm,流動相為0.01mol/L 磷酸二氫鈉水溶液(pH調至3.5)和甲醇以體積比85∶15 混合的溶液,流速1mL/min。進樣量20μL。采用式(2)、式(3)計算載藥材料的藥物包封率()和載藥量()。
將載藥材料與其他輔料分別過80 目篩,按表1 處方量混勻后,進行粉末直接壓片。片重(200±3)mg。采用三組不同接枝率的PVA--mPEG樣品以及原PVA、mPEG 重復以上操作,總共制得5組片劑。其中mPEG 由于溶解性差異(溶于丙酮)選擇熔融分散法載藥,即先將鹽酸二甲雙胍均勻分散于熔融的mPEG 中,干燥后再粉碎壓片。

表1 鹽酸二甲雙胍片劑處方
1.4 分析測試方法
1.4.1 傅里葉紅外光譜(FTIR)
通過傅里葉紅外光譜儀的ATR 附件進行了原料、mPEG-Cl 中間體和PVA--mPEG 樣品的紅外檢測。掃描范圍為500~4000cm,分辨率4cm,掃描次數32 次,使用OMNIC 8.3 軟件對所有光譜進行背景吸光度校正。
1.4.2 核磁共振氫譜(H NMR)
采用超導核磁共振波譜儀記錄原料PVA、mPEG 和PVA--mPEG 樣品的質子核磁共振光譜,以氘代二甲基亞砜(DMSO)為溶劑,四甲基硅烷(TMS)為內標。
1.4.3 差示掃描量熱法(DSC)
采用差熱示差掃描量熱儀研究了原料PVA、mPEG、二者物理混合物(質量比1∶1) 和PVA--mPEG 樣品的熱性能。升溫速率10℃/min,測試溫度0~250 ℃。
1.4.4 X射線衍射(XRD)
采用固定靶X射線衍射儀檢測PVA和PVA-mPEG的XRD圖譜。X射線在40kV和40mA的銅靶下產生,掃描角度為5°~70°,掃描速度為10°/min。
1.4.5 溶脹性能
1.4.6 防濫用性能(水中藥物的提取率)
本研究主要考察材料在水中的防濫用性能,將鹽酸二甲雙胍片置于500ml 純水中,在37℃、100r/min 下提取藥物,分別于20min、40min、60min 時取樣,同時補充等量同溫介質,采用HPLC法檢測藥物提取量。
1.4.7 藥物體外釋放
在符合《中國藥典(2015 版)》要求的溶出儀中,采用轉籃法100r/min進行鹽酸二甲雙胍片的體外釋放研究,溶出介質分別為500mL的pH 1.2鹽酸溶液和pH 6.8 磷酸鹽緩沖液,檢測溫度為37℃。每隔一定時間進行取樣,同時補充等量同溫的溶出介質。濾液經微孔濾膜處理,采用HPLC 法在233nm波長處測定鹽酸二甲雙胍的含量。
2 結果與討論
PVA--mPEG 接枝聚合物的合成主要分兩步進行,如圖1所示。首先,修飾mPEG 的末端單羥基:采用三氟化硼乙醚絡合物BF-EtO 作為路易斯酸催化劑,環氧氯丙烷ECH 質子化作用產生的氧鎓離子與mPEG 的末端羥基進行反應,得到mPEG末端羥基被置換為氯代醇醚結構的關鍵中間體mPEG-Cl,為接枝反應提供反應性功能基。優選該法制備mPEG-Cl,反應在熔融體系中進行,避免使用有機溶劑,工藝簡單且成本低。由于mPEG與mPEG-Cl溶解性能相似,傳統的分離方法使得二者難以完全分離,而mPEG的增溶作用會促進PVA 的分散,因此在后續接枝物的純化過程中再分離未修飾成功的mPEG。然后在NaOH 溶液的堿性條件下,mPEG-Cl 中間體的氯代醇醚結構與PVA 的羥基發生醚化接枝反應制備PVA--mPEG接枝聚合物,并利用乙醇抽提純化接枝物。
2.1 ECH投料量對mPEG-Cl合成反應的影響
鑒于Lin 等修飾的PEG 分子量較低,本研究選用的mPEG 分子量相對較大,因此著力研究了ECH 與mPEG 的投料摩爾比對合成mPEG-Cl 的影響,結果如圖2 所示。結果表明,隨著(ECH)∶(mPEG)比例的增加,mPEG 的羥基轉化率先增加隨后出現飽和。這可以用濃度效應來解釋:起初過量的ECH 可以增加反應概率,有利于提高反應的轉化率。但隨著投料比的進一步增加,mPEG的濃度相對降低,轉化率并沒有隨之繼續增加,為節約成本,選擇(ECH)∶(mPEG)=1∶4 為最佳投料比。
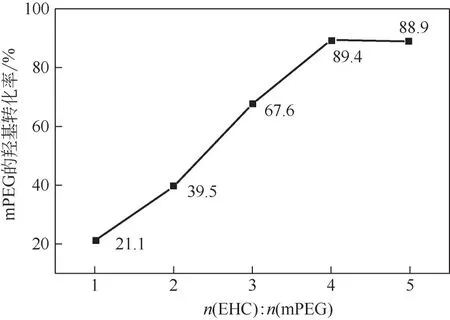
圖2 不同ECH物質的量投料比對mPEG-Cl合成的影響
2.2 溫度對合成PVA-g-mPEG接枝率的影響
由于mPEG-Cl 中間體與PVA 的醚化接枝對于反應溫度比較敏感,本文著力研究了反應溫度對合成PVA--mPEG 接枝率的影響,關系曲線如圖3所示。結果表明,PVA--mPEG 的接枝率隨著反應溫度的升高先升高再降低,并在80℃出現峰值,接枝率最高為120.48%。當反應溫度較低時,分子活化能較高,參與反應的碳氯鍵不容易斷裂,導致醚化程度較低;而當反應溫度過高時,堿性條件下的PVA 更容易發生皂化反應產生高溫凝膠化,致使參與反應的羥基數目大大降低,影響醚化接枝反應。綜上,最佳反應溫度為80℃。
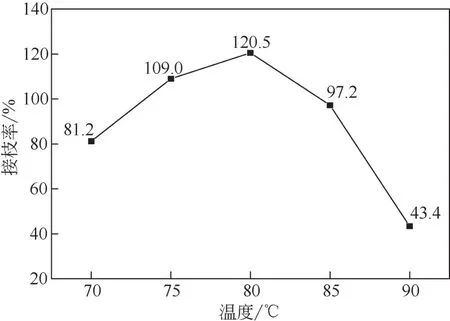
圖3 不同反應溫度對PVA-g-mPEG接枝聚合物接枝率的影響
將在不同溫度(90℃、70℃、80℃)下反應得到的三組不同接枝率的接枝聚合物分別命名為PVA--mPEG-1、PVA--mPEG-2、PVA--mPEG-3。
2.3 PVA-g-mPEG的結構表征
原料mPEG、PVA、中間體mPEG-Cl以及接枝聚合物PVA--mPEG 的紅外光譜如圖4 所示。由圖可見,中間體mPEG-Cl 的譜圖在波數740cm處出現了C—Cl 鍵的特征峰,由于反應屬于端基反應,C—Cl 鍵的含量較少,譜圖中C—Cl 鍵的振動強度也較弱。在PVA--mPEG 接枝聚合物的紅外光譜中可觀察到原料PVA 和mPEG 的主要特征帶。PVA--mPEG 接枝聚合物在3326cm處出現了羥基的伸縮振動峰,在1095cm處非常強的特征峰則歸因于醚鍵C—O—C的伸縮振動,表明mPEG接枝到PVA 主鏈上。同時,mPEG-Cl 的C—Cl 鍵特征峰在接枝物中未出現,表明接枝聚合反應中C—Cl基團已被完全消耗掉。因此,進一步證實了mPEG通過化學反應與PVA共價連接起來。
杜思雨想了老婆的種種好處,心里就越發不是滋味。都是這根該死的長頭發!可這根光潔美麗的長發絲從哪來的?又是誰的呢?又是怎么沾到自己的毛衣上的呢?杜思雨的酒此時已完全醒了,他在努力地回憶著,思索著。

圖4 原料及合成產物的FTIR譜圖
三組不同接枝率的PVA--mPEG 接枝聚合物的H NMR光譜如圖5所示。接枝聚合物在PVA(a=CH,b=OH,c=CH)和mPEG-Cl(d=CH,e=CH)處都有主要的質子峰,表明接枝反應成功,這與FTIR 分析一致。此外發現隨著接枝率的增加,側鏈mPEG上的單甲氧基上的質子峰信號也在不斷增強,表明有更多的mPEG接枝到主鏈PVA上。
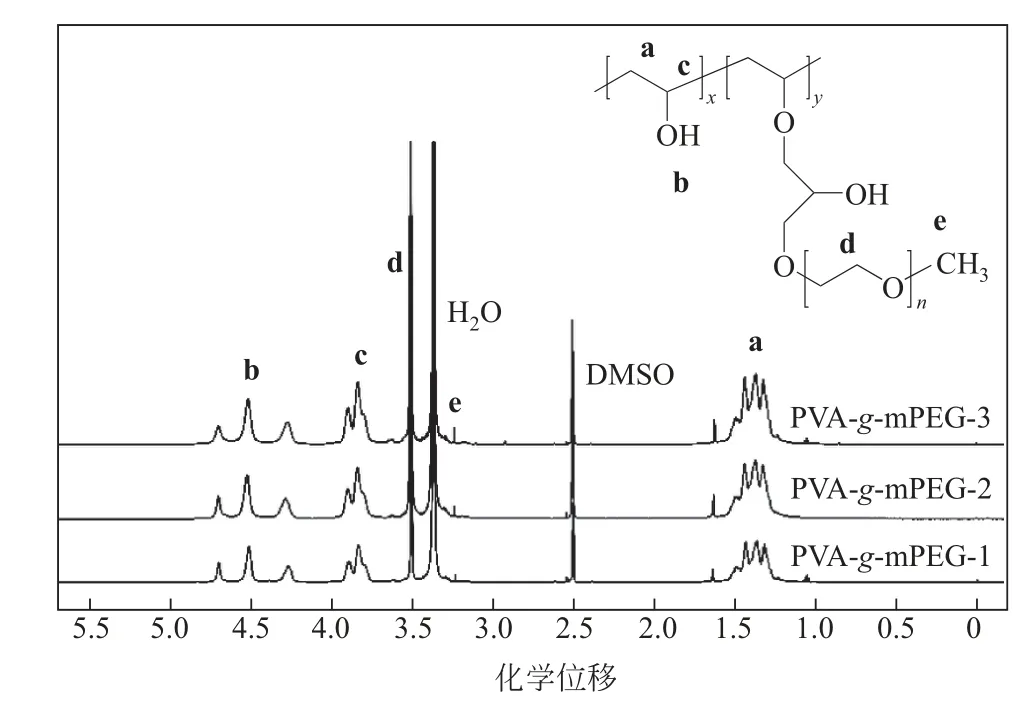
圖5 PVA-g-mPEG的1H NMR譜圖
2.4 DSC分析
圖6 顯示了原料PVA、mPEG、二者物理混合物以及三組不同接枝率的PVA--mPEG 接枝聚合物的DSC圖譜,可見原PVA和mPEG的熔融峰溫度分別為194.5℃和57.5℃;而采用溶劑共混法制備的PVA/mPEG 共混物的熔融峰溫度分別在194.5℃和57.2℃,與兩個原料的熔融峰有所對應;接枝物的mPEG熔融峰溫度均低于純mPEG,同時峰形變鈍,與簡單的物理混合物有明顯的不同,進一步表明了mPEG是通過化學鍵連接在PVA主鏈上,因此在熔融過程中會受到主鏈的束縛。隨著接枝率的升高,結晶熔融溫度升高,這是因為PVA含量相對降低,束縛作用減弱;PVA 熔融峰溫度升高則是其結晶更加完善所致,mPEG的引入使得PVA鏈段運動阻力減小而有利于其結晶。另外,由于本研究采用稱重法計算接枝率,接枝率的大小反映了接枝物體系mPEG含量的多少,通過化學鍵固定在PVA主鏈上的mPEG其實只占PVA重復單元的很少一部分,其對主鏈PVA 的結構改變相差較小,由此不同組成的接枝物中PVA的結晶熔融峰變化不大。
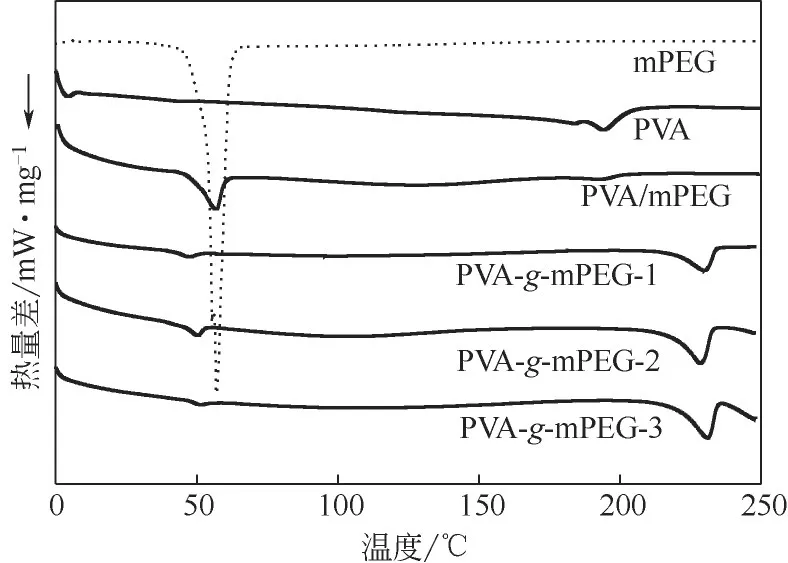
圖6 原料、物理混合物及合成產物的DSC譜圖
2.5 XRD分析
圖7 為原料PVA 以及三組不同接枝率的PVA--mPEG接枝聚合物的XRD圖譜。由圖可見,PVA在2為19.58°處出現較為尖銳的衍射峰,反映了半結晶PVA的(101)晶面;在2為11.63°、22.63°和40.64°處出現較弱的衍射峰。而PVA--mPEG接枝聚合物的圖譜顯示,相比于原PVA,主晶相的衍射峰變得更為尖銳,表明接枝聚合物的結晶度有所增加。這是由于mPEG具有增塑作用,側鏈的引入使得PVA鏈段運動阻力減小,有利于PVA鏈段規整排列從而使其結晶更加完善。這與PVA--mPEG接枝聚合物的DSC分析結果相吻合。
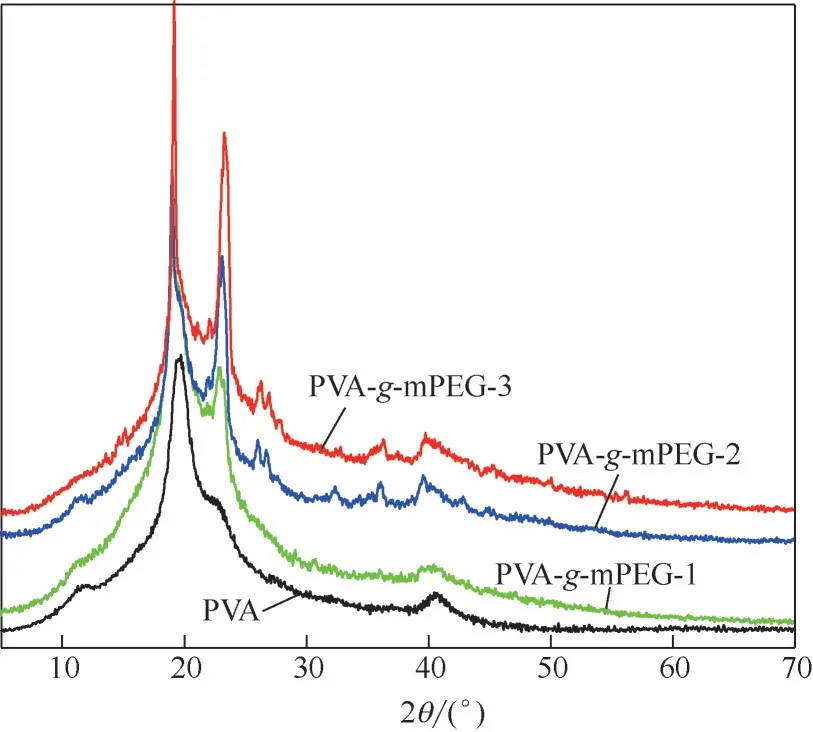
圖7 原PVA及合成產物的XRD譜圖
2.6 PVA-g-mPEG的溶脹性能
三組不同接枝率的PVA--mPEG 聚合物凝膠在水中的溶脹曲線如圖8 所示。由圖可知,PVA--mPEG系列接枝物都具有良好的溶脹性能,并且凝膠的溶脹率隨著側鏈mPEG含量的增加,平衡溶脹率呈現減小的趨勢。雖然原PVA 是一種親水性聚合物,分子鏈上含有大量的羥基,羥基可以與水分子發生氫鍵締合作用從而結合大量水分子,在冷水和熱水中均能較快地溶脹、溶解。但是,PVA--mPEG 接枝聚合物中PVA 結晶度隨著側鏈mPEG的引入而增加,同時因結晶形成的物理交聯點數量也隨之增加;前者致PVA 鏈段親水性能下降,后者使得“交聯”結構更為致密;兩者均使得吸水溶脹受限,進而導致平衡溶脹率有所降低。

圖8 PVA-g-mPEG接枝聚合物在水中的溶脹曲線
2.7 PVA-g-mPEG載藥及防濫用評價
由于阿片類藥物受到管控,選擇了一種高水溶性的活性藥物鹽酸二甲雙胍作為模型藥物,評價了PVA--mPEG 接枝聚合物的載藥效果和防濫用效果(藥物在水中的提取率)。由于PVA--mPEG接枝聚合物不溶于大部分有機溶劑,選擇二甲基亞砜作為良溶劑溶解接枝物與藥物,選擇丙酮作為不良溶劑,將藥物成功負載到接枝聚合物凝膠里,制備了3種不同接枝率的載藥聚合物凝膠樣品,載藥凝膠的藥物包封率及載藥量的計算結果列于表2中。結果發現3種不同接枝率的載藥凝膠的包封率及載藥量差別不大,但相對于原PVA都有所增加。結合PVA 熔融峰溫度有所升高,分析可能的原因是側鏈mPEG的引入使得PVA結晶更加完善,結晶形成了物理交聯點而更容易產生凝膠化,由此增加了載藥量和包封率。
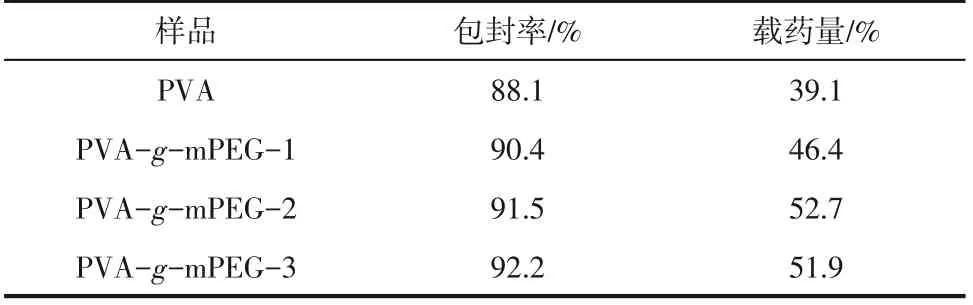
表2 不同基質載藥材料的藥物包封率及載藥量
藥物濫用者可通過水中提取藥物后靜脈注射實現濫用,將5組片劑分別置于500mL純水中進行藥物提取,評估了PVA--mPEG 的防濫用潛力,結果見圖9。結果表明,對于PVA 與mPEG 兩組對照片劑來說,20min 內藥物在水中的提取率分別為58.9%與97.9%,且PVA 組片劑在40min 時藥物可被提取87.6%。而三組樣品片劑在水中的藥物提取率都很低(小于30%),20min 時僅有28.3%、20.8%和12.9%的藥物可分別從PVA--mPEG-1、PVA-mPEG-2 和PVA--mPEG-3 三 組片劑中被提取。隨著時間的進一步延長,有65.3%、44.0%和38.8%的藥物在60min 后分別從三組樣品片劑中提取出來。這些結果表明,PVA--mPEG 接枝聚合物這一材料可以適度抵抗通過靜脈途徑的藥物濫用,因為只有一部分藥物可以通過傳統方法被提取,主要是因為當PVA--mPEG 載藥聚合物與水接觸時,可以形成黏性凝膠,進而導致注射器針頭堵塞以阻止靜脈注射濫用。隨著接枝率的升高,接枝物顯示出更強的防濫用特性。這可能是因為隨著接枝物支鏈數量的增加,載藥凝膠的親水性降低,被包裹其中的藥物也隨之更難釋放;也可能是因為隨著接枝率的升高,載藥凝膠的黏性也有所增大,也會阻礙藥物在水中的提取。
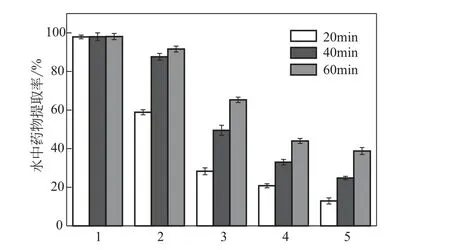
圖9 不同載藥基質的鹽酸二甲雙胍片在水中的藥物提取率
2.8 PVA-g-mPEG負載藥物體外釋放
分別在500mL pH 1.2的鹽酸溶液和pH 6.8的磷酸鹽緩沖液中進行了5組片劑的體外溶出研究,藥物釋放曲線如圖10所示。結果顯示,PVA與mPEG兩組對照片劑基本在40min時就釋放完全,由于二者均是親水性聚合物,在水中可以快速溶解,因此藥物很容易釋放。而對于三組不同接枝率的樣品片劑,發現在酸性介質中釋放2h 時,隨著接枝率的升高,藥物釋放有所降低,分別為96.0%、75.4%和29.1%。表明當接枝率達到一定程度時,聚合物材料有一定抵抗胃酸的能力,適合于對胃部刺激較大的阿片類藥物。而在pH 6.8磷酸鹽緩沖液中,兩組對照片劑基本也在40min時釋放完全。三組樣品片劑相對于對照片劑來說出現了不同程度的緩釋效果,以PVA--mPEG-2 組為例,藥物在3h 時才釋放80%以上。產生這一現象的原因可能是,當片劑與水接觸時,接枝聚合物快速水化溶脹,基體形成凝膠,藥物溶解后通過聚合物溶脹形成的通道逐漸擴散出去。另一方面,隨著接枝率的升高,聚合物分子量逐漸增加導致凝膠侵蝕減慢,藥物釋放行為相對延長,同時由于載藥凝膠所捕獲的藥物也更多,釋放量也隨之有所減少。
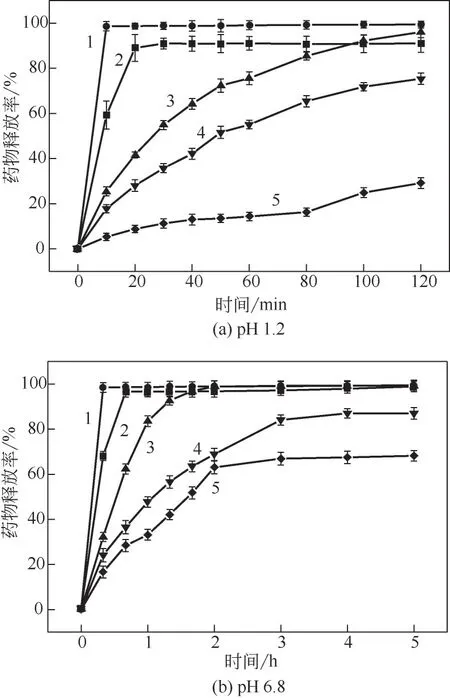
圖10 不同載藥基質的鹽酸二甲雙胍片分別在pH 1.2及pH 6.8溶液中的藥物釋放
3 結論
(1)以PVA、mPEG 和ECH 為原料,先修飾mPEG的末端單羥基得到氯代醇醚結構,再與PVA上的羥基發生醚化反應合成PVA--mPEG 接枝聚合物,接枝率最高可達120.48%。反應溫度在80℃以下時,接枝率隨溫度的升高而增加,進一步升高溫度致使接枝率不升反降的主要可能原因是PVA的高溫凝膠化。
(2)支鏈mPEG的引入具有增塑作用而有利于PVA 鏈段規整排列結晶;接枝聚合物溶液因此結晶構成的物理交聯點而凝膠化,致使接枝聚合物具有防濫用特性,且藥物包封率及載藥量相對于原PVA有所增加。
(3)以水溶性藥物鹽酸二甲雙胍為模型,隨著接枝率的升高(43.44%,81.23%,120.48%),20min 時水中藥物提取率降低(28.3%、20.8%,12.9%),接枝聚合物的防濫用特性增強,揭示了這種新型的PVA--mPEG 接枝聚合物作為藥用輔料在類似鹽酸基阿片類藥物防濫用制劑中的應用潛力。
符號說明
——載藥材料的載藥量,%
——載藥材料的藥物包封率,%
——PVA--mPEG接枝聚合物的接枝率,%
,m——PVA--mPEG 接枝聚合物的初始質量和時間時溶脹的質量,g
,——PVA 初始質量和PVA--mPEG 接枝聚合物的質量,g
——PVA--mPEG 接枝聚合物在水中的溶脹率,%
,,——載藥材料含藥量、投藥量和載藥材料的質量,g
