帶有蛋白質輸入的RNA-蛋白質結合位點預測方法
2022-03-18 05:01:08何如吉
計算機應用與軟件 2022年3期
梅 杰 何如吉 呂 強
(蘇州大學計算機科學與技術學院 江蘇 蘇州 215006)(江蘇省計算機信息處理技術重點實驗室 江蘇 蘇州 215006)
0 引 言
RNA和RNA結合蛋白(RNA Binding Proteins,RBP)的交互作用是理解轉錄后調控機制的關鍵,對蛋白質合成、基因融合和可變mRNA加工具有廣泛的影響[1-3]。RNA-蛋白質結合位點預測是指僅以RNA作為模型輸入,并為每一個RBP訓練一個模型用于預測RBP是否結合于輸入的RNA。得益于高通量測序技術的高速發展如CLIP-Seq[4],數以百計的RBP對應的大量RNA-蛋白質結合位點得以發現[5-8]。因此,通過機器學習方法預測RNA上的RNA-蛋白質結合位點成為了當前的研究熱點。其中深度學習相比傳統機器學習方法由于無需特征工程即可獲得良好的性能,近年來被廣泛應用到此問題上。
DeepBind第一個將卷積神經網絡(Convolutional Neural Network,CNN)用于提取RNA序列特征,在當時取得了突破性的進展[9]。隨后,沈紅斌課題組的系列模型(iDeep[10]、iDeepM[11]、iDeepA[12]、iDeepV[13]、iDeepE[14]、iDeepS[15]和CRIP[16])及Deepnet-rbp[17]、mmCNN[18]、CircSLNN[19]等模型運用深度學習方法對RNA-蛋白質結合位點預測問題進行了廣泛而深入的研究,包括長短時記憶網絡[20](Long Short-Term Memory,LSTM)、殘差神經網絡[21](Residual Network,ResNet)及注意力機制[22](Attention Mechanism)等方法都陸續被使用。盡管如此,這些方法都沒有考慮將RBP本身作為模型的輸入之一從而進一步擴大數據集并挖掘不同RNA-蛋白質結合位點問題的聯系。
從更高的視角來看,不同于RNA-蛋白質結合位點問題,RNA-蛋白質交互作用對問題同時需要RNA和RBP以一定形式作為輸入。由于同時獲取RNA和RBP數據的成本高昂,有限的數據量使得端到端的深度學習方法仍不能有效應用于這一問題[23]。而盡管高通量測序技術可以獲得單個RBP在特定細胞系和組織下的大量RNA-蛋白質結合位點,但將不同體內環境下的RNA-蛋白質結合位點進一步整合并構建更大的數據集可以進一步發揮深度學習模型的優勢。另一方面,模型通過對其他RNA-蛋白質結合位點數據的學習可能挖掘出與自身有關的知識。如在命名實體識別任務中,BioNER通過整合不同類型實體的數據集取得了性能的提升[24]。因此,本文提出一個整合不同CLIP數據的模型,并將RBP實驗編號以獨熱編碼的形式作為模型的輸入之一用來區別RNA序列將被哪個RBP結合。
在評估該模型效果時,將該模型在兩個RNA-蛋白質結合位點預測的權威數據集上與其他模型進行對比,結果表明該模型在這兩個數據集上相比其他模型均具有一定優勢。
1 算法介紹
本文提出的模型將不同RBP對應的實驗數據合并作為本模型的數據集,將RNA序列和RBP的實驗編號作為輸入并最終輸出對兩者結合概率的預測,模型結構如圖1所示。
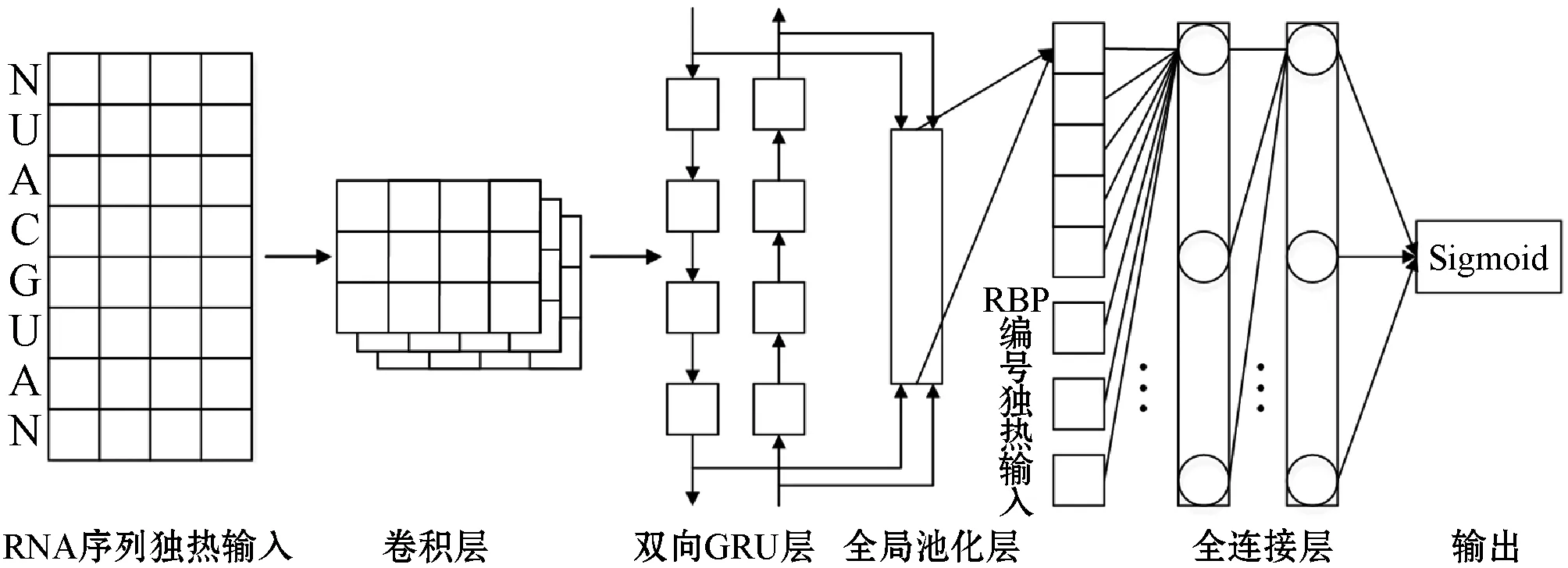
圖1 模型結構
RNA序列以獨熱編碼的形式表示,對于序列不等長的數據集,取訓練集中序列的最大長度n作為序列的輸入長度,并對長度不足的序列兩端以N補齊,其中N=[0.25,0.25,0.25,0.25]。RBP實驗編號的輸入向量寬度與訓練集中的實驗總數m一致,如RBP實驗編號為0的獨熱編碼表示為第0位為1而其他m-1位均為0的向量。
將RNA序列的獨熱編碼作為卷積層的輸入。第k個卷積核對齊到RNA序列位置i的輸出如式(1)所示。
(1)
式中:S是RNA序列的獨熱編碼表示,它是一個n×4的矩陣;Mk表示第k個卷積核的權重矩陣;b取值為1到4,表示A、U、C和G四種堿基,l表示卷積核長度;1≤i≤n-l+1且1≤k≤f,其中f指卷積核的數量。按以上步驟依次計算f個卷積核對RNA序列的輸出,則能得到一個大小為n×f的矩陣,即為CNN的輸出。一個卷積核就相當于一個特征選擇器,這里卷積運算用于學習RNA序列的局部特征,類比于圖像處理任務中的卷積運算得到圖像特征。
CNN層卷積處理后,應用修正線性單元(Rectified Linear Unit,ReLU)激活函數和批量歸一化層處理[25](Batch Normalization,BN)。ReLU可以對CNN的輸出進行非線性形變,批量歸一化層則可以加速模型收斂,并在一定程度上避免過擬合。
然后使用雙向門控神經單元[26](Bidirectional Gated Recurrent Unit,Bi-GRU)進一步提取RNA序列的全局特征。考慮到RBP對RNA序列的結合在生物學上并沒有一定的方向,所以這里使用雙向的設計。
一個GRU對于t時的輸入xt按式(2)-式(5)進行運算。而Bi-GRU包含正反兩個方向的GRU,它們在t時的輸出按式(6)合并。
zt=σ(Wz×[ht-1,xt])
(2)
rt=σ(Wr×[ht-1,xt])
(3)
(4)
(5)
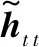
(6)

Bi-GRU的輸出經過全局最大池化層,得到了代表序列信息的特征向量。將特征向量與RBP實驗的獨熱編碼拼接起來,作為一個兩層均帶Dropout[27]的全連接層的輸入,它的輸出經過一個Sigmoid單元得到一個0到1之間的預測值,表示RNA序列在RBP實驗中的結合概率。
2 實驗與結果分析
為了評估本文模型的性能,本文選擇在RNA-蛋白質結合位點問題的兩個權威數據集(RBP-24和RBP-31)上與其他模型進行對比。
2.1 實驗數據與評價指標
RBP-24可在GraphProt[28]處下載,它由24個CLIP實驗組成并包含了21個不同的RBP。24個實驗中每個實驗的數據量不同,將24個實驗的訓練集合并并打亂后作為模型的訓練集,總計包含約120萬個樣本,訓練集序列的長度范圍在38個堿基對到375個堿基對之間。由于CLIP數據僅包含正樣本(即結合序列),GraphProt通過打亂結合序列順序的方式提供了數量相當的負序列。
RBP-31可在iONMF[29]處下載,它由31個CLIP實驗組成并包含了19個RBP。iONMF為每個CLIP實驗提供了劃分好的三組交叉驗證數據,每組數據的訓練集為30 000條,測試集為10 000條。iONMF選擇使用基因組上未被任何RBP結合的序列作為負序列,訓練集和測試集的正負樣本比例均為1 ∶4,序列長度為固定的101個堿基對。
兩組數據均以AUC(Area Under the ROC Curve)作為評價指標,見式(7)。
(7)
式中:M是正樣本的數量;N是負樣本的數量;positiveClass是正樣本的集合;通過對正樣本預測值進行排序,正樣本的最小預測值對應為rank1,以此類推ranki。
2.2 實驗環境與模型參數
實驗機器硬件配置為:CPU為兩塊Intel(R) Xeon(R) CPU E5-2620 v4 @ 2.10 GHz,GPU為三塊GeForce GTX 1080 Ti(每次訓練只使用一塊),內存大小為128 GB。模型是由Keras 2.2.2以TensorFlow 1.9.0為后端(backend)編程實現的。
本模型在RBP-24和RBP-31數據集上的主要參數設置分別如表1所示。權重和偏置均為Keras 2.2.2的默認設置。本模型的損失函數為交叉熵損失函數(CrossEntropy Loss),優化器選擇了Adam[30],學習率初始值是0.001。同時,本文還使用了早停技術及時中斷訓練,同時設置了檢查點來保存驗證損失最小的模型。

表1 模型主要超參數
2.3 RBP-24數據集上的實驗結果
本模型在RBP-24測試集上計算的AUC分布與其他模型對比的結果如圖2所示,對比模型的結果均來源于公開發表的論文。其中僅以序列作為輸入而不使用其他外源數據(如RNA的二/三級結構、region type和clip-cobinding等)的模型均以*標記,是對比的主要對象。特別要說明的是,Deepnet-rbp在原文中同時提供了僅以RNA序列作為輸入的結果,以及以RNA序列和RNA三級結構作為輸入的結果,本文使用的是前者。圖2中豎線表示模型在RBP-24測試集上24個RNA-蛋白質結合位點預測的AUC的平均值;圓圈為異常值,表示模型在某個測試集上的表現顯著低于其他測試集。
可以看出,本模型的預測結果整體分布較好,下界和均值分別是84.7%和93.9%,分別比iDeepE高出了8.9百分點和0.8百分點。
2.4 RBP-31數據集上的實驗結果
本模型在RBP-31測試集上計算的AUC分布和其他模型對比的結果如圖3所示。可以看到,使用傳統機器學習方法的GraphProt和iONMF盡管使用了RNA序列之外的數據源,相比深度學習方法仍然沒有優勢。而在所有僅以RNA序列作為輸入的模型中,本模型的平均AUC為87.3%排名第一,比DeeperBind[31]高出1.6百分點,甚至比額外使用了RNA結構信息的iDeepS還要高1.2百分點,與iDeepS的成對t-檢驗的單尾p-value遠小于1百分點,具有顯著差異性。而iDeep使用了RNA序列信息、結構信息、region type motif及clip-cobinding作為模型輸入,相比本文模型仍然有較大優勢。

圖3 不同模型在RBP-31測試集上的AUC分布
2.5 結果分析
從RBP-24和RBP-31的對比結果來看,本文模型相比現有的僅以RNA序列作為輸入的模型具有一定優勢。本文在處理RNA序列信息時使用了獨熱編碼+CNN+Bi-GRU的結構,這與DeeperBind的獨熱編碼+CNN+LSTM及iDeepS的獨熱編碼+CNN+Bi-LSTM均較為相似。但是,本文模型在RBP-31上卻取得了更好的結果,這說明了由于不同的RNA-蛋白質結合位點問題中存在著公共知識,具有生物學上的相關性,通過對其他RNA-蛋白質結合位點任務的學習確實帶來了目標任務的性能提升。同時,本文模型在參數規模上也有一定優勢。以iDeepS為例,它的卷積核數為16個,那么在RBP-31上的總卷積核數為496個,而本文模型僅使用了80個卷積核,這表明本文模型更有效地利用了模型參數。另外,從表1可以發現,盡管RBP-24和RBP-31在RNA序列長度上存在巨大差別,數據規模和RBP實驗數量也有不同,但是本文提出的模型卻可以以一套相同的超參數在兩個數據集上均取得出色的成績,這說明本文模型具有較強的泛化性能,不易過擬合,這也與訓練集的規模擴大有關。
3 結 語
本文提出的模型通過CNN-GRU結構提取RNA的序列特征,并通過將RBP的實驗編號以獨熱編碼的形式作為模型的另一輸入,擴展了模型訓練集的規模,深挖不同RNA-蛋白質結合位點問題的公共知識,進一步發揮了深度學習模型的優勢。在RNA-蛋白質結合位點預測任務中,本文模型在RBP-24和RBP-31這兩個數據集上均取得了比其他模型更好的結果。但是,如此大規模的訓練集對于RNA-蛋白質結合位點預測問題是一個相對陌生的領域,盡管使用CNN-GRU這一結構已經取得了一定進步,但是如何使用更復雜的技術、更深的網絡模型去充分挖掘數據中的信息仍然有進一步研究的空間。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
小獼猴智力畫刊(2022年9期)2022-11-04 02:31:42
中學生數理化·中考版(2022年11期)2022-02-16 07:01:20
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
小哥白尼(趣味科學)(2019年6期)2019-10-10 01:01:50
光學精密工程(2016年6期)2016-11-07 09:07:19
發明與創新(2016年38期)2016-08-22 03:02:52
太空探索(2016年5期)2016-07-12 15:17:55
核科學與工程(2015年4期)2015-09-26 11:59:03
