AT1R-Crim1信號通路在乳鼠肥大心室肌細胞Kir2.1mRNA和蛋白表達調控中的作用
2022-03-29 01:32:16霍照美楊英楊龍何炯紅夏桂玲郭楚嫻
貴州醫藥 2022年3期
霍照美 楊英 楊龍△ 何炯紅 夏桂玲 郭楚嫻
(1.貴州醫科大學,貴州 貴陽 550025;2.貴州省人民醫院心內科,貴州 貴陽 550002)
心肌異常肥大導致的心室重構是慢性充血性心力衰竭(CHF)發生、發展最主要的病理生理機制[1]。惡性室性心律失常導致的心臟性猝死是CHF患者主要的死亡原因[2]。心室肌細胞離子通道重構是導致細胞動作電位時程改變,進而引發惡性室性心律失常的重要病理生理學基礎[3]。調控心肌肥大的信號因子眾多,半胱氨酸豐富跨膜成骨蛋白調控因子(Crim1)作為血管緊張素受體1型(AT1R)下游信號通路參與大鼠心室肌細胞肥大的負性調控[4]。并且,Crim1過表達能夠抑制致肥大因子誘導的心室肌細胞膜瞬時外向鉀電流(Ito)的改變[5]。但是,目前關于AT1-Crim1信號通路參與肥大心室肌細胞離子通道重構的報道極少。內向整流鉀電流(Ik1)參與動作電位復極后期進程,對穩定心肌細胞靜息電位、保障心臟復極儲備具有重要作用。編碼Ik1離子通道α亞基的Kir2.1基因突變可導致長、短QT綜合征[6]。本研究旨在探討AT1-Crim1信號通路在肥大心室肌細胞Kir2.1 mRNA和蛋白表達中的作用。
1 材料與方法
1.1試劑與儀器 血管緊張素Ⅱ(AngⅡ)和氯沙坦(Los)購自Sigma公司。胰蛋白酶、Ⅱ型膠原酶、高糖DMEM培養基、特優級胎牛血清(FBS)、5-溴脫氧尿嘧啶核苷(5-BrdU)均為Gibco公司產品。兔抗鼠Crim1抗體購于北京博奧森公司。兔抗鼠Kir2.1抗體購自美國abcam公司。山羊抗兔FITC-IgG抗體、BCA蛋白質定量試劑盒購自北京中杉金橋公司。攜帶Crim1基因的重組腺病毒(Ad-Crim1)、腺病毒空載體(Ad-null)購自上海ThermoFisher SCIENTIFIC公司。TransStart?Top Green qPCR SuperMix、TransStart? First-Strand cDNA Synthesis SuperMix購自北京全式金生物技術有限公司。其余試劑均為進口分裝或國產分析純。PCR儀(MJ Research);熒光顯微鏡(Leica公司);Bio-Rad化學發光儀、Bio-Rad 550酶標儀(Bio-Rad公司)。
1.2實驗動物 清潔級SD大鼠1 d齡乳鼠40只,雌雄不限,由廣東省實驗動物學會提供,動物實驗證書編號15276。 本研究通過貴州省人民醫院倫理委員會審批,倫理審查批件編號(2020)076號。
1.3實驗方法
1.3.1乳鼠心室肌細胞分離與培養 分離乳鼠左心室,保留室間隔,酶解法分離細胞,差速貼壁結合BrdU抑制成纖維細胞而獲得純化心室肌細胞[5]。
1.3.2細胞干預分組 細胞培養24 h,更換無血清DMEM培養基,分組干預。干預試劑包括:腺病毒空載體(Ad-Null),Crim1基因重組腺病毒載體(Ad-Crim1),血管緊張素Ⅱ(AngⅡ,終濃度0.1 μM),氯沙坦(Los,終濃度10 μM)。對照組:加入MOI=50對應量的腺病毒空載體,感染6 h后換成2倍體積新鮮無血清無雙抗DMEM,繼續培養至48 h。AngⅡ組:加入MOI=50對應量的腺病毒空載體,感染6 h后換成2倍體積新鮮無血清無雙抗DMEM,感染24 h后加AngⅡ,繼續培養至48 h。Los+AngⅡ組:加入MOI=50對應量的腺病毒空載體,感染6 h后換成2倍體積新鮮無血清無雙抗DMEM,感染24 h后加AngⅡ、Los,繼續培養至48 h。Crim1組:加入MOI=50對應量的Ad-Crim1,感染6 h后換成2倍體積新鮮無血清無雙抗DMEM,繼續培養至48 h。Crim1+AngⅡ組:加入MOI=50對應的Ad-Crim1,感染6 h后換成2倍體積新鮮無血清無雙抗DMEM,轉染24 h后加AngⅡ,繼續培養至48 h。Crim1+Los+AngⅡ組:加入MOI=50對應的Ad-Criml,感染6 h后換成2倍體積新鮮無血清無雙抗DMEM,轉染24 h后加AngⅡ、Los,繼續培養至48 h。
1.3.3肥大刺激有效性鑒定 RT-qPCR測定心室肌細胞β-MHC的mRNA表達。結晶紫染色細胞,拍照,軟件測量細胞面積大小。
1.3.4RT-qPCR檢測mRNA水平 Trizol法提取細胞總mRNA,按TransStart?Top Green qPCR SuperMix、TransScript? First-Strand cDNA Synthesis SuperMix的說明書步驟檢測并計算各基因的mRNA相對表達量。引物序列見表1。

表1 各基因的引物序列
1.3.5Western蛋白質免疫印跡檢測 提取細胞總蛋白,測定蛋白濃度。取40 μg總蛋白上樣,電泳后轉移蛋白至NC膜上,5%脫脂牛奶封閉1 h。加兔抗大鼠GAPDH抗體(1:1 000),兔抗大鼠Kir2.1抗體(1:1 000)或兔抗大鼠Crim1抗體(1:1 000),4 ℃孵育過夜。充分漂洗后,加入辣根過氧化酶標記的Ⅱ抗室溫孵育1 h。充分漂洗后顯影、攝片并進行條帶灰度定量測定。

2 結 果
2.1各組乳鼠心室肌細胞β-MHC mRNA表達水平和細胞面積比較 各組間β-MHC mRNA表達水平總體比較差異有統計學意義(F=72.229,P<0.001);AngⅡ組和Crim1組β-MHC mRNA表達水平較對照組明顯升高(P均<0.001);Los+AngⅡ組(P=0.008)、Crim1+AngⅡ組(P<0.001)和Crim1+Los+AngⅡ組(P<0.001)β-MHC mRNA表達值皆較AngⅡ組明顯下降;Crim1+Los+AngⅡ組β-MHC mRNA表達值較Los+AngⅡ組明顯下降(P<0.001)。各組心室肌細胞表面積總體比較差異有統計學意義(F=75.730,P<0.001);AngⅡ組細胞表面積較對照組明顯增大(P<0.001);Los+AngⅡ組、Crim1+AngⅡ組和Crim1+Los+AngⅡ組細胞表面積皆較AngⅡ組明顯減小(P均<0.001);Crim1+Los+AngⅡ組細胞表面積較Los+AngⅡ組明顯下降(P=0.039)。見表2。
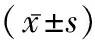
表2 各組心室肌細胞β-MHC mRNA表達水平和細胞表面積的比較
2.2Ad-Crim1、Los抑制AngⅡ刺激導致的心室肌細胞Crim1的mRNA和蛋白表達下調 各組心室肌細胞Crim1 mRNA表達值進行比較差異有統計學意義(F=710.507,P<0.001);AngⅡ組Crim1 mRNA表達水平較對照組明顯降低(P<0.001),而Crim1組較對照組明顯升高(P<0.001);Los+AngⅡ組(P=0.003)、Crim1+AngⅡ組(P<0.001)和Crim1+Los+AngⅡ組(P<0.001)Crim1 mRNA表達值皆較AngⅡ組明顯增加;Crim1+Los+AngⅡ組Crim1 mRNA表達值較Los+AngⅡ組明顯增加(P=0.035)。各組心室肌細胞Crim1蛋白表達值比較差異有統計學意義(F=198.967,P<0.001);AngⅡ組Crim1蛋白表達水平較對照組明顯降低(P<0.001),而Crim1組較對照組明顯升高(P<0.001);Los+AngⅡ組(P=0.008)、Crim1+AngⅡ組(P<0.001)和Crim1+Los+AngⅡ組(P<0.001)Crim1蛋白表達值皆較AngⅡ組明顯增加;Crim1+Los+AngⅡ組Crim1 mRNA表達值較Los+AngⅡ組明顯增加(P=0.005)。見表3。
表3 各組心室肌細胞Crim1和蛋白表達水平的比較
2.3Ad-Crim1、Los抑制AngⅡ刺激導致的心室肌細胞Kir2.1的mRNA和蛋白表達下調 各組心室肌細胞Kir2.1 mRNA表達值比較差異有統計學意義(F=53.823,P<0.001);AngⅡ組Kir2.1 mRNA表達水平較對照組明顯降低(P<0.001),而Crim1組較對照組明顯升高(P<0.001);Crim1+AngⅡ組和Crim1+Los+AngⅡ組Kir2.1 mRNA表達值皆較AngⅡ組明顯增加(P均<0.001);Crim1+Los+AngⅡ組Kir2.1 mRNA表達值較Los+AngⅡ組明顯增加(P=0.007)。各組心室肌細胞Kir2.1蛋白表達值進行比較差異有統計學意義(F=52.330,P<0.001);AngⅡ組Kir2.1蛋白表達水平較對照組明顯降低(P<0.001),而Crim1組較對照組明顯升高(P<0.001);Los+AngⅡ組(P=0.004)、Crim1+AngⅡ組(P<0.001)和Crim1+Los+AngⅡ組(P<0.001)Kir2.1蛋白表達值皆較AngⅡ組明顯增加;Crim1+Los+AngⅡ組Kir2.1蛋白表達值較Los+AngⅡ組明顯增加(P=0.004)。見表4。
表4 各組心室肌細胞Kir2.1 mRNA和蛋白表達水平的比較
3 討 論
心肌肥大、心衰患者室性心律失常風險顯著增加,其機制之一是存在心室電重構[7]。離子通道重構是這些心臟電生理變化的重要基礎。心肌肥大的發生機制涉及眾多信號轉導通路。骨形態發生蛋白(BMPs)屬于轉化生長因子-β(TGF-β)超家族成員。在壓力負荷和AngⅡ誘導的心肌肥大中BMP4表達上調,而BMP4的表達能誘導心肌細胞肥大、凋亡和心肌纖維化,并能增強AngⅡ所誘導的心肌肥大效應[8]。Crim1是一種在心臟高表達的胚胎基因[9]。它擁有vWF-C型富含半胱氨酸重復片段(CRR)的跨細胞膜結構[10],其結構類似于BMPs抑制劑Chordin,這樣的結構使其成為TGF-β亞家族的調控分子[11]。Crim1通過CRR片段與BMP4、BMP7結合,對BMPs起抑制作用[12]。Crim1廣泛存在于胚胎各組織器官,并影響這些器官的發育,包括調控心臟的發育[13]。
我們的前期研究[14]發現,在腹主動脈縮窄大鼠心室肥大模型,心室肌組織中Crim1蛋白表達下調,AT1R阻滯劑替米沙坦干預可抑制Crim1蛋白表達下調和心室肥大。在牽張刺激誘導的培養乳鼠肥大心室肌細胞模型,Crim1的mRNA和蛋白表達下調;AT1R特異性阻滯劑Los干預顯著抑制心室肌細胞肥大,并抑制致肥大因素誘導的Crim1 mRNA和蛋白表達下調[4]。通過攜帶Crim1基因的病毒感染促進培養心室肌細胞Crim1蛋白表達,明顯抑制機械力牽張和苯腎上腺素刺激的細胞肥大;并且明顯抑制苯腎上腺素刺激的心室肌細胞Ito的改變[5]。以上研究結果顯示,Crim1作為AT1R下游信號通路參與大鼠心室肌細胞肥大的負性調控,并可能參與心室肌細胞膜離子通道重構的調控。本研究通過致心肌細胞肥大因子AngⅡ刺激構建心室肌細胞肥大模型。給予AT1R特異性阻滯劑Los干預,能顯著抑制AngⅡ刺激誘導的心室肌細胞肥大效應及Crim1 mRNA和蛋白表達下調,并抑制AngⅡ刺激誘導的心室肌細胞Kir2.1 mRNA和蛋白表達下調。給予Ad-Crim1感染心室肌細胞促Crim1 mRNA過表達干預,顯著上調Crim1蛋白表達、下調肥大基因β-MHC的mRNA表達;并明顯抑制AngⅡ刺激誘導的心室肌細胞肥大及Kir2.1 mRNA和蛋白表達下調。Los和Ad-Crim1聯合干預較Ad-Crim1單獨干預能更顯著抑制AngⅡ刺激心室肌細胞誘導的上述效應。該結果顯示,Crim1作為AT1R下游因子參與心室肌細胞肥大及Kir2.1 mRNA和蛋白表達的調控,但其并非AT1R調控心室肌細胞肥大和Kir2.1表達的唯一的下游信號因子。
綜上所述,AT1R-Crim1信號通路參與AngⅡ誘導的乳鼠肥大心室肌細胞Kir2.1 mRNA和蛋白表達的調控。
