表面單分子量子態的探測和調控研究進展
2022-03-30 14:26:16姚杰趙愛迪
物理學報 2022年6期
關鍵詞:分子結構
姚杰 趙愛迪
1) (中國科學技術大學化學物理系,合肥微尺度物質科學國家研究中心,合肥 230026)
2) (上海科技大學物質科學與技術學院,上海 201210)
1 引言
分子尺度的材料和結構由于其內稟的量子化能級結構、特殊的波函數特征,展示出了許多新穎的電子學、光學和磁學等物理性質,并可用來制備豐富功能的器件,如單分子開關、導線、整流器、晶體管、存儲器和量子比特[1,2].無論是沿著經典比特的邏輯思路繼續研究分子尺度的量子器件,還是開辟全新的以量子比特為基礎的量子信息處理方式,都需要我們深刻理解分子尺度量子效應,發展新的量子設計方法與量子調控手段,構筑新的量子器件,最終實現信息的存儲、傳輸與處理等多種功能.其中,單分子尺度結構的量子態的探測和調控是首當其沖的核心問題.在這里,量子態為廣義的定義,即物質處于可由某一或若干量子數描述的狀態,這些量子態可以被用于經典的非相干信息處理,也可以進一步被調控和用于量子相干信息處理.分子因其原子數目遠小于固體,具有典型的分立的量子化能級,因此,對于分子尺度的物質而言,其量子態主要包括:分立能級的電子態(量子點的分立能級即與分子的類似);分立能級躍遷帶來的光子態;局域磁矩帶來的自旋態;其他的分立能態諸如振動態等也是量子化的.單分子尺度的量子態探測與調控的概念,起源于分子電子學研究熱潮的興起.在2000—2005 年左右,得益于掃描探針顯微學的發展,使得對表面吸附單分子量子態的探測成為可能,涌現出單分子自旋態探測、單分子發光等一批重要的研究成果.經過十多年的發展,這一領域雖然已經取得了長足的進步,但也遇到了相當大的瓶頸,面臨著諸多挑戰,基于分子的量子器件的實現仍然道阻且長.問題諸如:已有分子不能滿足實際應用的要求,需要對分子尺度量子結構進行新的設計和性能調控;基于分子的器件中分子與電極的連接不可或缺,所以需要深刻理解分子-電極之間、以及分子-環境之間的相互作用;分子量子器件在工作過程中必然伴隨電荷與能量的轉移,需要在分子尺度通過量子設計和調控,實現對相關電子態的激發、弛豫機制、壽命的準確了解和控制.在這篇綜述中,我們將主要以表面單分子為對象,對其光子態、自旋態、電子態等的探測、表征與調控的相關研究做一下回顧,并提出一些下一步研究的設想,希望能拋磚引玉,尋找下一個突破口.
單分子量子態的探測和調控,首先面臨的問題是研究手段的制約.對于單分子,通常附著于襯底,而掃描探針顯微鏡的發明,為探測單個分子帶來了極大的契機.1981 年由IBM 的Binning 和Rohrer等[3]根據量子隧穿原理發明了第一臺掃描隧道顯微鏡(scanning tunneling microscopy,STM)裝置.1983 年,他們首次利用STM 觀測到了Si(111)7×7重構表面的實空間原子成像[3].這也顯示出了STM 極高原子分辨能力的優越性,這一發現將物質的微觀狀態以及原子和分子的實空間分布首次直觀地展現在我們面前,使得原子和分子真實地被“看”到.之后在1990 年,IBM 實驗室的Eigler 和Schweizer[4]在Ni(110)表面,利用STM 操縱35 個Xe 原子,將無序的Xe 原子排列成規整的IBM 這3 個字母,由此開創了STM 強大的原子操縱能力.這一工作也為之后的原子操縱構建全新的人造量子體系[5-8]以及人工精準調控單分子的化學反應打開了大門,激發了人們對STM 原子操縱能力的研究興趣.另外在此基礎上,STM 還開發出了電導譜學功能,結合STM 的高分辨性能可以探測分子體系局域的電學性能,主要包括I-V,dI/dV,以及d2I/d2V.I-V譜可以研究單分子的輸運性質.而dI/dV譜以及圖反映了電子結構和局域態密度分布,可以用來研究分子的電子結構.d2I/d2V譜是根據電子的非彈性隧穿原理得到的,可以用來研究化學鍵的振動模等信息[9,10],以及自旋激發信息[11,12].例如Yu 等[13]就利用dI/dV研究了分子軌道的糾纏態,而Ho 等[14,15]則利用d2I/d2V譜分辨出了單個分子的內部化學鍵結構以及分子間成鍵的化學鍵結構.為了進一步豐富STM 的探測范圍以及提高STM 的分辨率,可以功能化STM 的針尖.例如在STM 尖端吸附1 個CO 分子,可以使得尖端更尖銳,提高STM 的分辨率,另外通過針尖吸附1 個磁性分子或者原子,可以實現對樣品磁性自旋的探測[16-19].例如最近Czap 等[11,12]通過在STM針尖吸附1 個磁性的二茂鎳分子,并用二茂鎳分子功能化的STM 針尖再去掃描二茂鎳單分子,實現了對二茂鎳單分子自旋振動激發態和自旋交換相互作用的探測.
上述研究表明,STM 已經成為了目前研究表面單分子量子態最強有力的工具.除了STM 外,目前對于單分子量子態的探測手段還有非接觸式的原子力顯微鏡(NC-AFM)[20-23]、太赫茲-掃描隧道顯微鏡(THz-STM)[24-28]、針尖增強拉曼光譜(TERS)[29-33]、STM 誘導發光技術(STML)[34-38]以及電子自旋共振(ESR)[39-41]等方法.而這些方法多在基于STM 技術上升級發展而來,需要與STM 一起聯用.目前研究分子尺度的量子態主要以金屬配位的酞菁分子和石墨烯納米帶這兩類體系為載體,本文主要介紹基于STM 技術這兩類體系中分子尺度量子態的探測和調控的研究進展,將先從表面單分子量子態體系的制備開始展開,再從單分子的光學性質和單分子的磁性自旋量子態以及最近興起的石墨烯類分子的拓撲態和自旋態三方面重點介紹單分子體系的量子態探測和調控.
2 表面單分子量子態體系的制備
單分子體系的量子態可從制備、調控、表征三個方面進行介紹,如圖1 所示,這里制備方法有STM 原位操縱和表面反應化學合成兩種,單分子量子態整體可分為電子態[13,42]、光子態[43]及自旋態[44,45]三種,且對其單分子量子態的調控也主要針對這三種,其電子態、光子態、自旋態可以通過STM,THz-STM,NC-AFM,ESR-STM,TESRS,STML 等手段進行表征.化學合成的分子在自由空間中本身就有可能具有優越的量子特性,但當分子置于表面上后,其量子態的保持和探測往往并不容易,這其中最大的問題就來自于襯底與其相互作用會改變分子的量子特性.因此,表面單分子量子態體系的有效制備和獲取是進一步探測和調控的前提.目前,制備合適的表面單分子量子態體系主要有原位操縱和表面化學合成兩大路線,分別對應于傳統材料的“自上而下”和“自下而上”制備方法.

圖1 表面單分子結構的量子態的制備、表征與調控的研究示意圖Fig.1.Schematic of the research on the synthesis,characterization and manipulation of the quantum states of surface-supported single molecular structures.
2.1 STM 原位操縱
STM 本質上是一種原位的近乎接觸式的探測手段,可以就近實現對分子的操縱.因此,在STM發明后的十多年里,人們已經發展出了多種原位操縱的手段.比如機械操縱,通過控制針尖來拖曳或推動分子.最近的研究已經可以通過機械操縱的方法制備出多自旋耦合的人造單分子結構[46,47],演示了量子態的靜態耦合.除了機械操縱外,通過針尖發射電子來“切割”和“焊接”分子中的特定基團,即電操縱,也已經被廣泛用于實現對分子的改性,進而實現對具有可觀測量子態的表面單分子結構的制備.2005 年,Zhao 等[48]利用針尖對吸附于Au(111)表面CoPc 分子進行“切割”,實現了其中Co 離子d 軌道中1/2 自旋態的恢復.CoPc 分子如圖2(a)所示,從dI/dV譜上可看出吸附在Au(111)表面的鈷酞菁分子沒有近藤效應(圖2(c)黑色線).用掃描隧道顯微鏡尖端的電壓脈沖從分子上切下8 個氫原子(圖2(b)),使該分子的4 個軌道與金基底發生化學鍵合.這種人工分子結構恢復了局域自旋,從dI/dV譜上可看出在費米表面附近觀察到了明顯的近藤共振(圖2(c) d-CoPc)[48].通過這種方法首次實現了對單個分子自旋態的調控.類似的方法Li 等[49]也在FePc 上實現,利用分子手術切除了FePc 8 個瓣上的H 原子.不同的是,在切除之前,在dI/dV譜上觀察到近藤共振,而切除最外層的8 個氫原子后,dI/dV譜特征變為雙臺階結構,反映了分子自旋態的非彈性轉變.

圖2 (a) CoPc 的結構模型,在實驗中,1 個瓣的氫原子2 和3 被解離;(b) STM 電流引起的脫氫示意圖;(c) 不同溫度下CoPc 和脫氫CoPc(d-CoPc)的dI/dV 譜;(d) STM 圖像顯示了連續尖端誘導的CoPc 在Au(111)上的脫氫[48]Fig.2.(a) Structural formula of the CoPc.Hydrogen atoms 2 and 3 of one lobe were dissociated in the experiments.(b) Diagram of the dehydrogenation induced by the STM current.(c) dI/dV spectra of CoPc and dehydrogenated CoPc (d-CoPc) at different temperatures.(d) STM images showing the sequential tip-induced dehydrogenation of a CoPc on Au(111)[48].
2.2 表面分子原位合成
STM 原位操縱雖然可以非常有目的地針對特定的局域化學鍵和基團進行操縱,但其效率很低,難以實現大規模的制備.2010 年左右,在表面直接合成分子(on-surface synthesis)的化學路線實現了重大的突破.Cai 等[50]利用“自下而上”的方法,通過小分子前驅體在金屬表面反應輔助條件下合成了石墨烯納米帶.這種方法利用設計好的分子前驅體在金屬單晶表面進行化學合成石墨烯分子結構,可以實現對石墨烯分子結構原子級別的精確控制.而且這種方法制備出的石墨烯分子結構質量高無雜質,有利于對石墨烯分子結構的性質探測.通過選取不同的前驅體,還可以合成不同類型的石墨烯分子結構.此方法中,首先通過設計和合成前驅體小分子,然后將前驅體小分子通過熱蒸發的方式蒸鍍到金屬表面,例如Au(111)或Ag(111)表面.退火到一定溫度,經過熱活化后脫鹵和烏爾曼偶聯等反應后環化脫氫(cyclodehydrogenation)形成聚合的石墨烯分子納米結構,用設計好的分子前驅體來進行精確控制“自下而上”地生長[51-58].圖3 展示了合成具有不同手性石墨烯納米帶(chGNRs)的具體方法,包括所使用的前驅體小分子、合成路線,以及產物的STM 圖像[59].
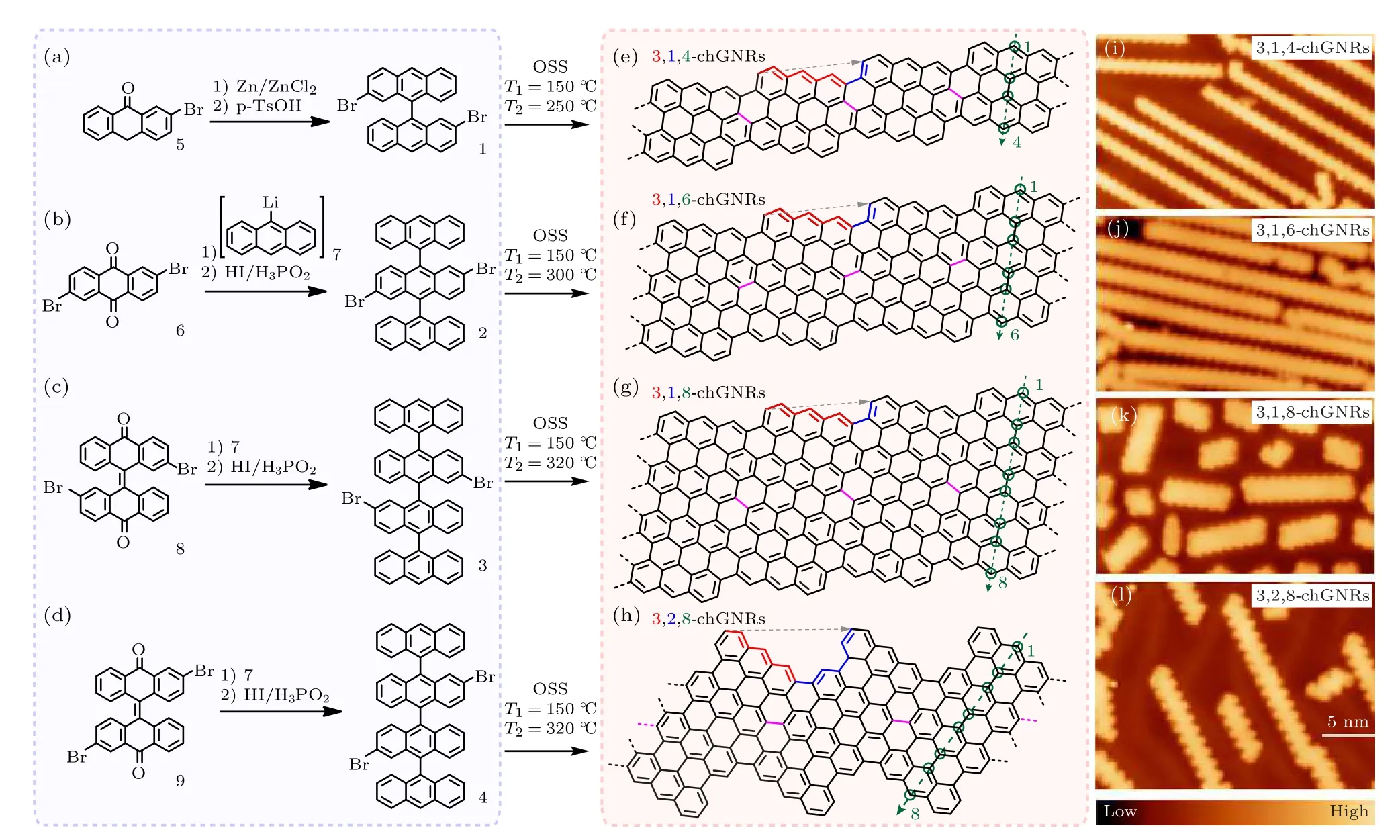
圖3 結合溶液和表面合成chGNRs 的方法 (a)—(d) 溶液法合成不同原子寬度3,1,w-chGNRs 的分子前驅體1,2,3 和3,2,8-chGNRs 的前驅體4;(e)—(h) 利用4 種分子前驅體分別靶向chGNRs 的化學結構;(i)—(l) 在Au(111)表面合成chGNRs 的STM 圖像[59]Fig.3.Synthetic strategy to produce chGNRs combining solution and on-surface synthesis:(a)—(d) Solution synthesis protocols for producing molecular precursors 1,2,3,for the synthesis of 3,1,w-chGNRs with different widths,and precursor 4 for 3,2,8-chGNRs;(e) —(h) targeted chemical structures of chGNRs by using the four molecular precursors in (a) —(d),respectively;(i)—(l) STM overview images of the chGNRs formed on a Au(111) surface[59].
3 單分子光子態的探測和表征
單分子光子態的探測和表征主要指利用單分子的量子特性使其作為單光子源的探測和表征研究.單分子具有分立的能級、確定的幾何構型,這些與量子點都極為相似,而且單分子還天然具有全同性,使其發光特性具有極佳的一致性.單光子源毫無疑問是構建量子計算、量子密碼、量子存儲以及量子信息通信的核心[60-67].如何制備可控穩定高效的單光子源一直是量子光學追求的重要目標之一.相比于稀土離子[68,69]、量子點[70-72]、色心體系[73]的單光子源,單分子體系的單光子源發光頻率豐富、量子產率高、室溫操作且易于集成.早在2000 年Lounis 和Moerner[60]就利用脈沖激光激發固體中單個分子實現了可控的單光子源.這種方法觸發單個光子產生的速率很高,與其他單光子源相比,具有簡單、室溫操作且可升級空間大的優勢.因此單分子發光體系非常有潛力用來構建穩定可控且高效的單光子源.而基于STM 電流誘導的單分子發光,可以將STM 的空間分辨以及強大的對分子的操縱能力運用到誘導分子發光中,是到目前為止單分子發光研究的最主要手段.
在這里我們重點介紹運用掃描隧道顯微鏡實現的電致發光和光致發光.在用STM 對單分子進行發光研究的時候,一般常選擇金、銀、銅作為基本襯底,同時為了防止熒光淬滅,還需要讓金屬襯底和單分子脫耦合.實驗中最常用的方法是在基本金屬襯底上再通過熱蒸發的方式生長NaCl 薄膜作為脫耦合的襯底,NaCl 薄膜的層數可以根據實驗的需要自由控制.
3.1 單分子電致發光
目前對于單分子電致發光的研究主要有分為兩種途徑,一種是基于微納加工,采用微納電極直接對單分子施加電流作用從而使其發光[74].另一種是采用STM 技術,用隧穿電子誘導單分子發光[75,76].而鑒于STM 的實空間分辨能力以及其獨特的可以對單分子進行操縱的能力,采用STM誘導的電致發光,更能從根本上研究單分子電致發光的機理.例如Svec 研究小組[77,78]采用CO 功能化STM 針尖后同時進行了針尖增強光子探測和高分辨率原子力成像,將NC-AFM,STM 及單分子電致發光有機結合,以及通過STM 對CuPc 分子的操縱,使其位于Na+或Cl—離子的中心,進而操控單個CuPc 分子的雙激子態的能量變化.這些實驗研究都展現了STM 對于研究單分子電致發光的獨特優勢.
電致發光機制中一個重要的現象為上轉換發光.上轉換電致發光是一種發射的光子能量高于激發電子能量的現象,其在激光信息技術、紅外探測、生物醫學等領域有巨大的應用前景.雖然在STM 隧道結內的上轉換電致發光早期研究中已經提出了代表性機制,包括分子間三重態湮滅[79-81]和分子振動輔助等離激元泵浦[34,82].然而,早期的這些觀察都是在分子多層膜上進行的,阻礙了競爭機制的潛在分化和主導機制的識別.2019 年Chen等[83]通過在銀襯底上利用NaCl 薄膜作為脫耦合層,在其上沉積了酞菁分子,并結合STM 系統構建出酞菁單分子電致發光體系(如圖4(d)).該工作在單分子水平上觀測到電致上轉換發光現象,有力排除了此前對這類體系中上轉換機制的一些猜測.Qx(1.81 eV 單線態)峰值的強度在不同的偏壓區呈現出3 個明顯的逐步增加,表明不同的物理過程占主導地位.以此為基礎的理論分析進一步表明,兩種不同的分子激發機制——非彈性電子散射和載流子注入以及之間的微妙競爭導致了不同偏壓區域不同的發光現象.基于對上述兩種激發機制以及對系統其他潛在激發態的定量分析,首次提出了以自旋三重態作為中間過渡態,同時結合非彈性電子散射和載流子注入兩種激發過程的單分子上轉換發光機制[83].緊接著,他們又在多個單分子相干偶極體系中,發現了單光子的超輻射.在Ag(100)襯底上,利用NaCl 薄膜作為脫耦合層,同時利用STM 的單分子操縱能力,構建了由從2 到12 個非鍵合的鋅酞菁分子鏈(如圖4(a)),并且研究了這些分子鏈體系通過局域隧穿電子激發的發光特性以及隨鏈長變化發光特性的演化[84].隨著分子單體數目的增加,超輻射模式的峰位紅移,且峰寬變窄.能量分辨的光子圖可以反映出不同排布方式的偶極相互作用信息.對分子鏈的超輻射模式進行空間上的量子產率表征,可得到超輻射模式的光子圖.共線同相的超輻射模式的能量分辨光子圖表現出中間暗兩邊亮的啞鈴形狀圖案(圖4(b)).這種只有中間1 個節點的圖案,說明Chen 等[84]構造的分子鏈在被電子激發后形成的是1 個耦合體系,測量的超輻射模式對應的離域激子態存在于整個分子鏈上.更為有意思的是,在分子鏈的輻射最強點測量了輻射光子的二階相干函數,發現超輻射具有單光子特性(如圖4(c)).這也證明整個分子鏈處于單激子態,必須被當作單一的量子體系來看待.當分子鏈體系中的1 個分子被局域電子激發后,激發能會迅速地離域到整個分子鏈,形成單激子超輻射態,進而產生單光子超輻射現象.該實驗研究揭示了分子集體態的光學性質及其與分子尺度局域等離激元的相互作用.另外分子鏈超輻射模式的單光子輻射特性使得其可以作為單光子光源應用在量子計算和量子存儲中[83].這一研究成果表明少數分子的聚集體也有望和單分子一樣作為單量子體系用于量子光學等應用中.

圖4 (a) 誘導分子鏈發光的STML 示意圖[84];(b) 多達12 個單體ZnPc 分子鏈的STM 形貌圖,不同分子鏈的典型STML 譜,ZnPc 鏈超輻射態的實驗光子圖像[84];(c) 不同長度的分子鏈光子發射的二階相關函數測量結果[84];(d) STM 誘導的單分子發光示意圖,右邊是分子結構的俯視圖;不同偏置電壓下單個H2Pc 分子的電致發光光譜;恒定電流下Qx 峰的歸一化偏置電壓和光強依賴關系圖,對數圖在插圖中[83].Fig.4.(a) Schematic of STML from ZnPc molecular chains[84].(b) STM images of ZnPc molecular chains of up to 12 monomers,typical STML spectrum of different molecular chains,experimental photon images for the superradiant states of the ZnPc chains[84].(c) Second-order correlation functions g (2)(τ) for different ZnPc chains[84].(d) Schematic of the STM-induced single-molecule emission.A top view of the molecular structure is given on the right.Electroluminescence spectra from a single H2Pc molecule at different bias voltages.Normalized bias-dependent intensity of the Qx peak at a constant current,with the logarithmic plot shown in the inset[83].
3.2 單分子光致發光
盡管STM 誘導的單分子電致發光是一種獨特而強大的研究光激發的技術,但它在激發過程中難以精確地選擇性激發特定的分子能級.與光譜學中使用的可調諧激光器相比,隧穿電子的能量不是非常明確而且其能量也并非單色,這使得利用STM隧穿電流選擇性地激發單個量子態變得特別困難.這阻礙了對單個激發態內在特征(如能級和線寬)的解釋,以及對后續動態過程中由一種態到另一種態轉變過程的描述.如果能利用STM 實現光致發光,將能使光致發光的分辨率提高到新的臺階.Yang 等[85]通過使用經過特殊修飾后的STM 針尖和金屬襯底構造出1 個納腔,并用激光激發其內部的等離激元場(圖5(a)),高度局域的等離激元場使得單分子發射光子并且再次與等離激元場耦合至遠場.ZnPc 單分子的光子圖中呈現出明顯的中間暗,而在4 個葉瓣上亮度有顯著增強(圖5(b)),這說明了電偶極模型的失效,需要將電子態躍遷密度也考慮進來.在光子圖中沿著AB虛線做一條高度曲線,其中光子圖的空間分辨率達到了驚人的8 ?(圖5(c))[85].2021 年Imada 等[86]通過使用可調諧的激光代替固定能量的激光用于STM誘導單分子發光中(圖5(d)),發現其發光強度得到了大幅提高.這種可調諧的單色納米探測可以實現對單分子的單個電子和振動量子態的能級和線寬進行狀態選擇性表征.STM 的熒光光譜表明,納腔等離激元的局部電磁場強烈增強光致熒光的過程,所有光致熒光峰均源自被測的單個分子.即使在非常弱的1 μW 激發功率下,總檢測光子強度(能量積分)也超過180000 光子/秒,這表明其效率比尖端增強拉曼光譜和使用固定能量激光的光致熒光光譜高出幾個數量級(如圖5(e)).
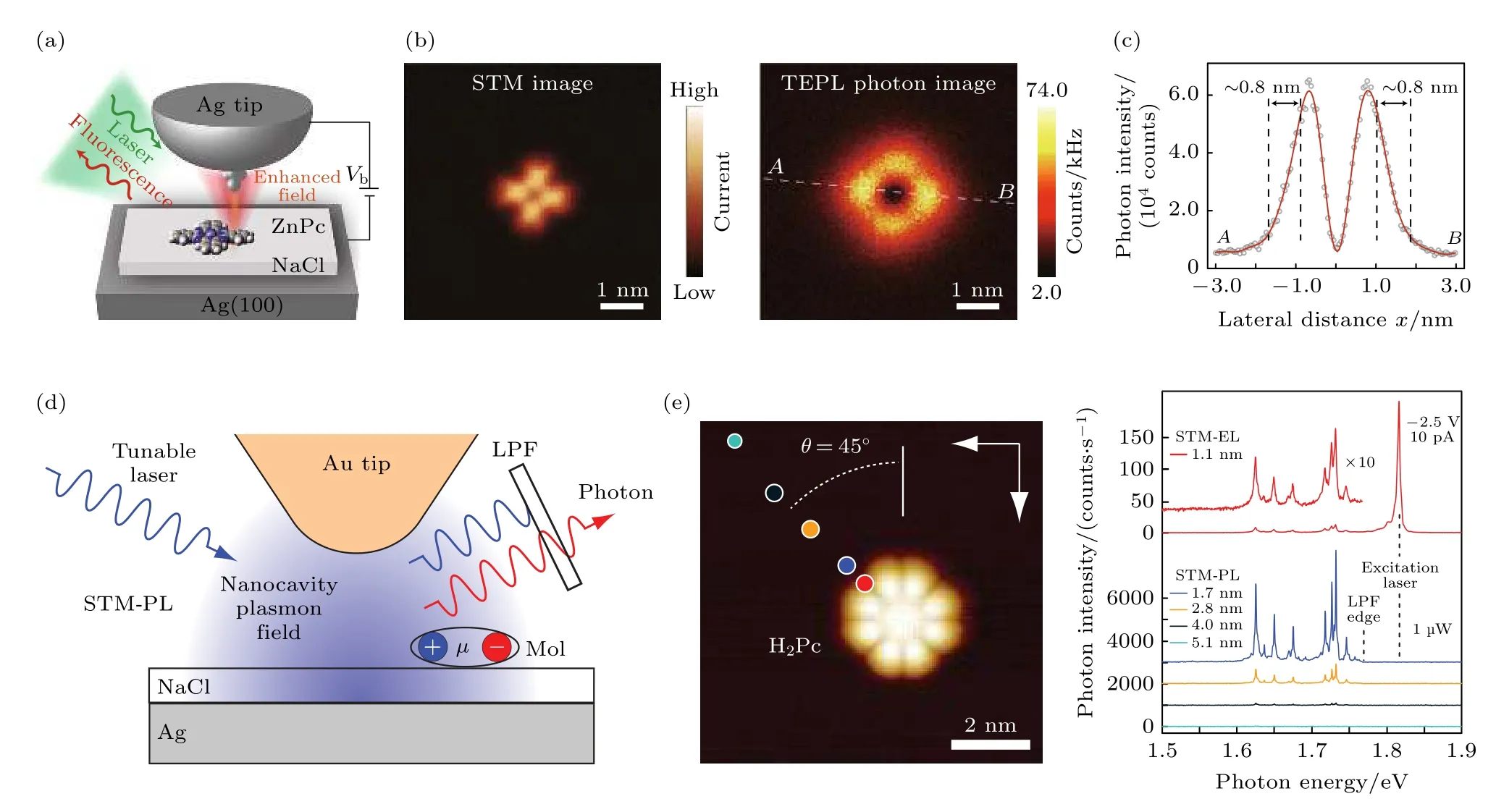
圖5 (a) 針尖增強光致發光實驗模型[85];(b) ZnPC 分子的STM 圖(左)和光子圖(右)[85];(c) 在光子圖像(b)中虛白線AB 的光子強度側面圖[85];(d) STM-PL 實驗示意圖[86];(e) H2Pc 分子的放大 STM 圖,STM 圖(左)中的圓圈顯示的是光譜測量時針尖的位置,其顏色與相應光譜的顏色匹配[86].Fig.5.(a) Schematic of the experimental of Sub-nanometre-resolved single-molecule TEPL[85];(b) simultaneously recorded STM image (left) and TEPL photon image (right) of a single ZnPc molecule [85];(c) photon intensity profile for the dashed white line AB in the photon image in b (right)[85];(d) schematic depiction of STM-PL measurement[86];(e) a magnified STM image of a H2Pc molecule,and the measurement tip positions for the spectra shown in STM image (left)are indicated with circles whose color matches that of the corresponding spectrum[86].
4 單分子局域自旋態的探測和調控
近年來單分子在納米器件和自旋電子學中的潛在應用得到了廣泛的研究.許多實驗已經證明可以利用分子構成基本器件,包括負微分電阻隧道結[87]、整流器[88,89]、放大器[2]和數據存儲[90].通過操縱單個磁性分子或原子的自旋自由度來進行對信息的編碼是分子原子尺度電子學持續發展的核心挑戰之一[91].而要想將其真正投入應用,那么關鍵的問題是對單分子的自旋態進行有效地控制.對表面單分子系統和分子水平的相關研究,不僅能從根本上理解分子系統的磁自旋特性,而且對于實現其實際應用具有特殊意義.盡管目前已經有多種檢測和控制單分子自旋的方法,例如光學檢測磁共振技術[92-94]、磁共振力顯微鏡[95-97]、氮-空位(NV)色心磁力計[98]和基于斷裂結的分子裝置[99]等.但這些系統的分辨率還不足以到單分子和單化學鍵的尺度,難以實現對分子內和分子間自旋-自旋相互作用的探測.STM 通過測量分子的微分電導(dI/dV)譜(即STS)來檢測表面分子的局域磁性,可以實現精準地探測單分子間和單分子內部的局域自旋態.具體而言,對于表面吸附的磁性分子系統,STS 測量經常觀察到近藤共振和自旋激發誘導非彈性電子隧穿的特征峰,并將其作為分析自旋信息的關鍵證據.近藤共振是費米能級附近的譜異常現象,起源于磁性分子的局域自旋與襯底傳導電子之間的交換相互作用.因此,近藤效應通常在分子自旋載流子和導電襯底之間存在較強相互作用的系統中被觀察到,如最近She 等[100]發現在半金屬表面吸附的CoPc 分子中的單自旋能夠得到很好的保持而不會淬滅.由于外部磁場或自旋-軌道相互作用,具有分裂自旋態的磁性分子,其自旋激發可以誘導非彈性電子隧穿,其dI/dV譜在費米能級處會表現出對稱的階梯狀特征[44].另外通過STM 與電子自旋共振譜(ESR)的結合可以實現在成像的同時連貫地控制表面上單個原子的自旋.最近利用此技術研究了單配位配合物-鐵酞菁(FePc),并研究了分子間自旋(FePc-FePc 二聚體)以及分子與原子間自旋(FePc-Ti)之間的磁性相互作用.并發現FePc 的分子自旋密度既分布于中心Fe 原子,又分布于配體(Pc),從而產生了強烈的分子幾何結構依賴的交換耦合[39].在實現表面單分子局域自旋態制備和探測的基礎上,如何對其進行調控是當前這一方向的研究重點.這其中,針尖作為不可或缺的一部分,可以通過施加局域電場來操縱分子的自旋態,也可以通過操縱原子實現自旋態的改變,還可以通過調控針尖物態和隧道結特性來實現對分子自旋態的調控.
4.1 針尖局域電場調控
2013 年,Liu 等[101]報道了一種可逆地調控單分子自旋態的方法.將MnPc 單分子吸附在Au(111)上,MnPc 分子表現出突出的交叉特征(圖6(a)).通入H2導致原始MnPc 分子轉化為中心有凹陷的結構(圖6(b)).通入H2引起的MnPc 分子中Mn離子對H 原子的化學吸附,從而轉化為H-MnPc.此外還發現,通過在H-MnPc 分子上施加正的尖端脈沖可以導致H 的分離,讓H-MnPc 分子再次轉化成MnPc 分子.H 原子對MnPc 的化學吸附也導致dI/dV測量值發生顯著變化.MnPc 在Au(111)上的dI/dV譜在零偏壓處呈現階梯狀特征(圖6(c)中的紅色曲線),且在施加磁場時可以使階梯狀特征發生分裂,因此可以推斷出這種零偏壓異常是由于近藤共振導致的(圖6(d)).相比之下,H-MnPc 的dI/dV曲線無特征峰(圖6(c)中的藍色曲線).通過通入H2實現MnPc 到H-MnPc的轉換,以及通過針尖脈沖使得H-MnPc 上脫H 重新變成MnPc,可以實現dI/dV譜上近藤特征曲線與無特征曲線之間的可逆轉換(圖6(c)).計算結果表明,與MnPc 相比,H-MnPc 中Mn 離子的d 電子數幾乎不變,但有效電荷在d 軌道上重新分布,導致分子的凈自旋從MnPc 的S=3/2 降低到H-MnPc 的S=1.此外,H 附著也導致Mn 與襯底更大程度地分離,從而削弱了Mn 與襯底耦合,這兩個因素都有助于抑制H-MnPc 中的近藤效應[101].通過H 原子的化學吸附可以可逆調節MnPc 在Au(111)上的自旋態,這也為調控表面吸附單分子的自旋提供了思路,從而制備出具備量子自旋的單分子體系.
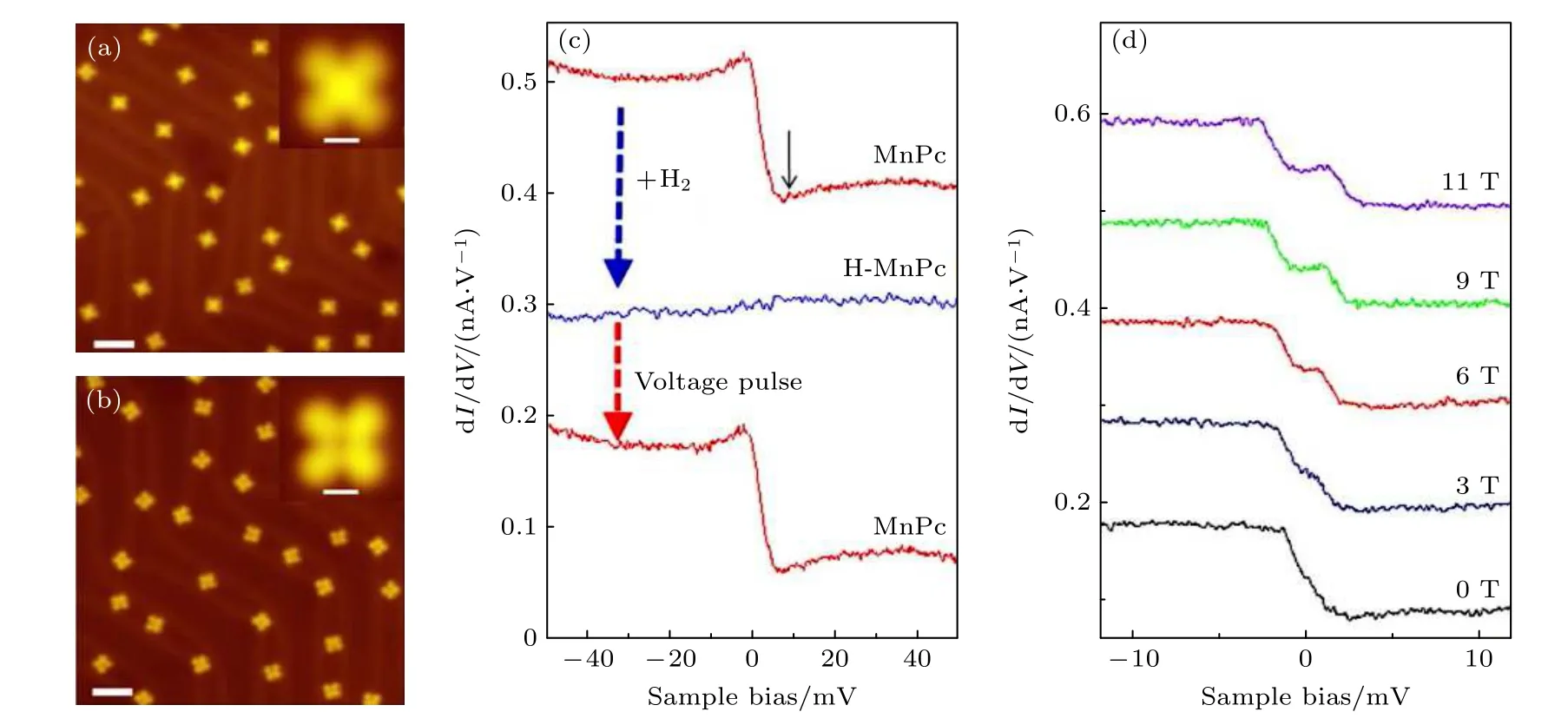
圖6 Au(111)上 (a) MnPc 和 (b) H-MnPc 的STM 圖像;(c) 由H 原子吸附和解吸引起的MnPc 分子中心記錄的dI/dV 譜的連續變化;(d) MnPc 近藤特征在磁場下的演化[101]Fig.6.STM images of (a) MnPc and (b) H-MnPc on Au(111);(c) sequential variations of dI/dV spectra recorded at the center of a MnPc molecule induced by the adsorption and desorption of a H atom;(d) magnetic-field evolution of the Kondo feature of MnPc[101].
而同樣利用針尖局域電場進行對分子磁性自旋的操縱,不同于Liu 等[101]的工作,Komeda 等[102]通過針尖脈沖控制分子旋轉從而實現對自旋態的調控.dI/dV測量揭示了近藤效應特征的位點依賴性:近藤特征峰在分子的葉瓣周圍很突出,但在中心處零偏壓對應的dI/dV強度減弱(圖7(a)).這表明近藤共振由Pc 配體上的自旋負責.然后,Komeda 等發現當通過對分子施加尖端脈沖來旋轉上部Pc 配體時,可以實現可控地打開和關閉近藤效應.當TbPc2分子的2 個Pc 配體之間的方位角(θ)為 45° 時,其中心在STM 圖像中顯得更亮(圖7(b)),可檢測到近藤特征峰(圖7(d) 中的頂部曲線).打脈沖后,分子的中心變暗,對應的TbPc2分子中2 個Pc 配體之間的方位角θ變化到了 30°(圖7(b)).同時,dI/dV測量顯示分子的近藤共振消失(圖7(c) 中的底部曲線).近藤效應的這種依賴于夾角θ的轉換被解釋為分子自旋狀態變化的結果.對于θ=45°的TbPc2分子,吸附在Au(111)上的分子的Pc 配體具有源自未配對π 電子的S=1/2 自旋.然而上配體旋轉到θ=30°導致前沿分子軌道的重排,這使得電荷從表面轉移,淬滅分子自旋,從而淬滅近藤狀態.通過施加電流脈沖可以實現在2 個穩定的配體取向之間進行可逆切換,這能夠實現在單分子水平上自旋態信息的編碼[102].

圖7 (a) 在 Au(111) 上的 TbPc2 分子的葉瓣上和中心處記錄的 dI/dV 譜.插圖:組裝結構中 TbPc2 分子的 STM 圖像.(b) θ=45°和θ=30°的TbPc2 分子的示意圖和STM 圖像.(c) 在應用尖端脈沖之前和之后在 TbPc2 分子處獲得的 dI/dV 譜[102].Fig.7.(a) dI/dV spectra recorded at the lobe and center of a TbPc2 molecule on Au(111).Inset:STM image of TbPc2 molecules in the assembled structure.(b) Schematic illustrations and STM images of TbPc2 molecules with θ=45° and θ=30°.(c) dI/dV spectra acquired at a TbPc2 molecule before and after the application of a tip pulse[102].
4.2 金屬原子摻雜調控
雖然一些不含過渡金屬的自由基分子本身即可以具有單自旋態[103],但如何能夠調控一個無過渡金屬的非自由基分子使其產生局域自旋是一個值得探索的方向.對于過渡金屬參與的金屬有機配合物,如金屬酞菁(Pc)分子,因為d 電子的存在通常具有順磁性.Pc 是一種廣泛使用的大環螯合配體[104],Pc 與金屬原子的配位通常是通過吲哚環的—NH—基團中的2 個H 原子的離解后發生的,導致Pc 配體的—2 價態.因此,穩定的金屬酞菁(MPc,M=Mn[44],Fe[49],Co[48],Cu[105])分子更喜歡在Pc 和可以穩定在+2 價態的金屬(例如3d 金屬)之間形成.作為比較,無d 電子的主族金屬Al(+3)并不易與PC 形成配合物.Hong 等[106]采用真空合成的方法,通過H2Pc 分子和Al 單質在 Au(111) 表面的共沉積,實現了對H2Pc 分子的金屬化,制備出了AlPc 分子.圖8(a)是H2Pc 和AlPc單分子的STM 圖以及它們的結構模型.分別對這兩種分子做dI/dV譜可以看出它們的電子態結構.在AlPc 葉瓣(圖8(b)中的最頂部曲線)獲得的dI/dV譜中檢測到零偏置峰,證明了AlPc 中自旋的存在.該分子的自旋態密度起源于未配對π 電子,該電子主要在Pc 環的瓣上離域.這解釋了AlPc中心沒有近藤共振(圖8(b)中的中間曲線).氯化的AlPc (ClAlPc)是一種由Al 原子與Cl 原子軸向鍵合形成的AlPc 衍生物,在Au(111)上的進一步比較研究(圖8(c))表明,吸收Cl 原子后形成ClAlPc 分子沒有近藤特征(圖9(d)),因為氯化后ClAlPc 的所有分子軌道都被雙重占據.因此,AlPc 在Au(111)上的自旋態可以通過Cl 原子與Al 中心的連接/分離來調節.這種方法也為調控無過渡金屬分子的磁性提供了新的思路,即通過摻雜主族原子(如這里的Al 和Cl)實現對分子磁性自旋態的兩級調控.
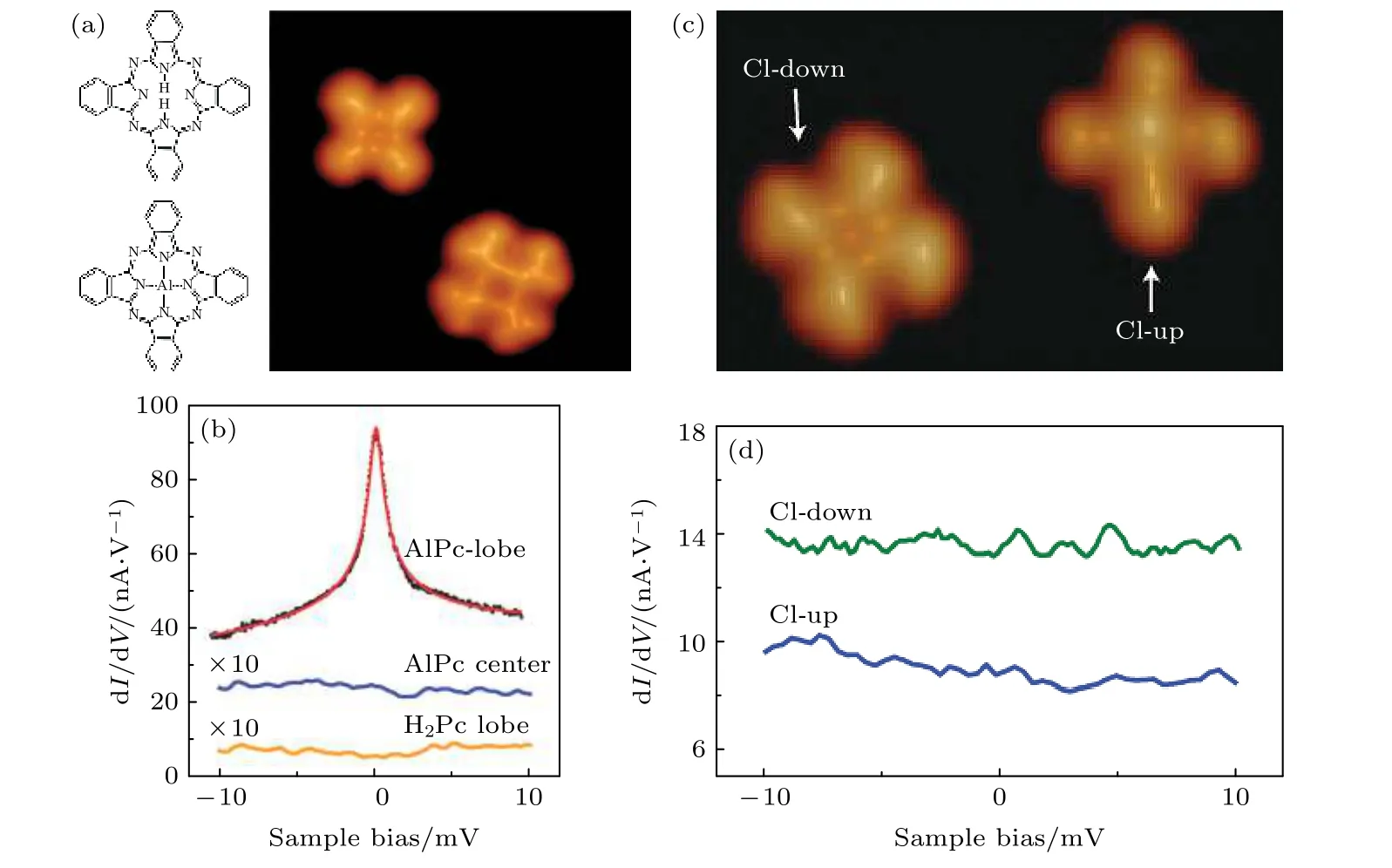
圖8 (a) H2Pc 和Al 共沉積后的2 個分子的化學結構模型和STM 圖.STM 圖左上角和右下角的分子分別是H2Pc 和AlPc.(b) 將尖端置于AlPc 瓣(黑色)、AlPc 的Al 中心(藍色)和H2Pc 瓣上方(黃色)得到的譜.(c) ClAlPc 的 STM 形貌圖.(d) ClAlPc瓣的 dI/dV 譜.藍色:Cl 向上,綠色:Cl 向下[106].Fig.8.(a) Chemical structure model and STM map of two molecules after co-deposition of H2Pc and Al.The top-left and bottomright molecules of the STM image are H2Pc and AlPc,respectively.(b) Spectra taken with the tip placed above a lobe of AlPc(black dots),the Al center of AlPc (blue),and a lobe of H2Pc (yellow).(c) STM topograph of ClAlPc.(d) dI/dV spectra of a lobe of ClAlPc.Blue:Cl-up,green:Cl-down[106].
4.3 隧道結調控
2017 年,Ormaza 等[107]通過在STM 金屬針尖上吸附1 個二茂鎳分子,構建出了金屬針尖-單分子-金屬襯底的單分子隧道結,可以實現單個分子自旋的可控轉換.當改變從隧道區到接觸區2 個電極之間的距離時,二茂鎳分子從自旋S=1 可逆地切換到S=1/2.在可重復的電導測量中觀察到的非彈性和彈性自旋翻轉機制在實驗上證明了這種切換,并通過第一性原理計算得到印證[107].
最近,Xing 等[108]也利用類似的方法構建出了單分子隧道結,在低溫下通過簡單地調節探針-樣品距離來調節隧道耦合,實現了近藤共振峰和非彈性電子隧道譜之間的可逆切換.首先使用超導Nb 針尖區分吸附在 Au(111)上的 FePc 分子的兩種不同構型(圖9(a)和圖9(b)).從形貌上看,它們表現出15°的旋轉角度差,在用普通W 探針采集的微分電導(dI/dV)譜中,構型II FePc 在費米面附近出現急劇下降,這是由于其近藤效應導致.相比之下,構型I FePc 表現出在費米面附近出現一對對稱臺階的特征,這些臺階特征來自于非彈性電子隧穿過程(圖9(c)).利用超導Nb 針尖研究吸附在Au(111)上的FePc 分子,dI/dV譜顯示了它們在費米能級附近都有1 個U 型能隙,該能隙歸因于Nb 尖端的s 波超導能隙.同時除了針尖提供的額外超導能隙特征外,譜的臺階邊緣也比W 針尖收集的更尖銳,這表明Nb 針尖的dI/dV譜能量分辨率得到增強(圖9(d)).通過STM 針尖拾取1 個FePC 分子的方法,將隧穿結的結構從Nb 針尖-絕緣體-FePc-金襯底(圖9 e)轉變為Nb 針尖-FePc-絕緣體-金襯底結構(圖9(f)),超導能隙內出現了類似Yu-Shiba-Rusinov 的特征(圖9(f)),這是分子的磁性自旋態與Nb 針尖的超導態相互作用的典型特征.該研究表明,調整隧道勢壘是一種有效的簡單方法,可以在單分子隧道結中實現原位可逆的自旋開關[108].

圖9 (a) 和 (b) 為組態I 和II 的 FePc 分子STM 圖像,分別顯示組態II 的“交叉”相對于組態I 的分子中心旋轉15° ;(c) 通過正常W 端在構型I 和II FePc 分子上獲得的dI/dV 譜,顯示分子2 種構型的電子狀態顯著不同;(d)通過超導Nb 針尖在構型I 和II FePc 分子上獲得的dI/dV 譜;(e) Nb-絕緣體-FePc-Au 隧道結結構以及其典型的dI/dV 譜,顯示了Kondo 特征峰;(f) Nb-FePc-絕緣體-Au 隧道結結構,以及其典型的dI/dV 譜,顯示了2 個間隙內YSR 態[108]Fig.9.(a) and (b) Typical STM images of configuration I and II FePc molecules,respectively,showing the “cross” of configuration II rotates with respect to the molecular center by 15° compared with configuration I;(c) dI/dV spectra obtained on configuration I and II FePc molecules by a normal W tip,showing strikingly different electron states for the two configurations of the molecule;(d) dI/dV spectra obtained on configuration I and II FePc molecules by a superconducting Nb tip;(e) typical dI/dV spectra in a Nb-insulator-FePc-Au tunneling junction,showing a Kondo dip;(f) typical dI/dV spectra in a Nb-FePc-insulator-Au tunneling junction,showing two in-gap YSR states[108].
5 石墨烯分子結構的量子態探測和調控
石墨烯是由排列成蜂窩狀晶格的單層碳原子組成,由于晶格對稱性,導帶和價帶在狄拉克點接觸并具有線性色散關系.這一不尋常的電子結構使得石墨烯有望用于設計新范式特性的器件.最近興起的轉角雙層石墨烯中關聯電子效應的研究更是給石墨烯的電子學器件研究帶來了新的方向.然而,單層石墨烯由于缺乏能隙,在電子器件中的應用受到限制.打開其帶隙的有效方法是將石墨烯的橫向尺寸減小到納米級,變成石墨烯納米帶或者石墨烯納米環等,通過量子限制和邊緣效應引入帶隙[109-111].這些石墨烯分子結構,本質上也可看成一種大分子,因此對其研究的方法和調控的手段也與單分子類似.石墨烯分子結構根據其邊緣結構的不同,大致可分為扶手椅邊緣和鋸齒狀邊緣[112-114].其中扶手椅邊緣的石墨烯分子結構表現出量子限制誘導的帶隙,且其帶隙依賴于帶狀寬度,這對其在電子學器件中的應用至關重要[115,116].相比之下,鋸齒形邊緣的石墨烯分子結構被預測具有自旋極化邊緣態的未配對π 電子,其電子庫侖斥力可以導致形成具有能隙的自旋極化電子結構.此外,這些鋸齒狀邊緣的石墨烯分子結構還表現出幾何和尺寸依賴的自旋輸運性質[117,118].
在2.2 節中,我們可以看到,表面原位合成的方法已經使得制備各種不同寬度、不同異質結構、不同邊緣結構的石墨烯分子結構成為可能,極大地拓展了單分子物態研究的范疇.近年來的研究表明,這些具有不同邊緣、不同寬度、不同幾何構型的石墨烯分子結構,可以表現出異常豐富的物性.其中最引人注目的是拓撲電子態和碳電子磁性.拓撲電子態本身并不一定是量子化的,然而對于拓撲絕緣體等體系的研究已經表明,拓撲電子態的實現和界面調控有望被用于實現量子化的局域態并用于量子計算.而石墨烯分子結構中的碳電子,則可以通過結構設計來實現無過渡金屬原子的磁性,使得這些帶有局域自旋態的石墨烯分子結構和磁性金屬原子、NV 色心等單自旋體系一樣有望用于量子信息,也有望用于自旋濾波器、自旋閥和自旋開關等自旋電子學應用[119].石墨烯分子結構的出現打開了石墨烯作為晶體管應用的大門,同時也豐富了石墨烯的用途,以及利用更加豐富的手段對石墨烯的各種物性進行調控[120].
5.1 石墨烯分子結構的拓撲電子態
隨著石墨烯納米帶(GNR)拓撲邊界態理論的提出,不同寬度、不同邊緣和不同末端的半導體石墨烯納米帶屬于不同的電子拓撲類.且石墨烯納米帶的拓撲相受空間對稱性的保護,并由邊緣的化學結構決定.不同拓撲相的2 個石墨烯納米帶之間的局域異質結電子態可以通過異質結邊界幾何結構來調節[121].如表1 所列,不同的邊緣結構和不同寬度都會對拓撲不變數產生影響.這使得石墨烯納米帶的拓撲電子態具有很高的可調性,即通過化學方法設計和制備多樣的幾何結構,可以有效地實現對其拓撲電子態的調控.
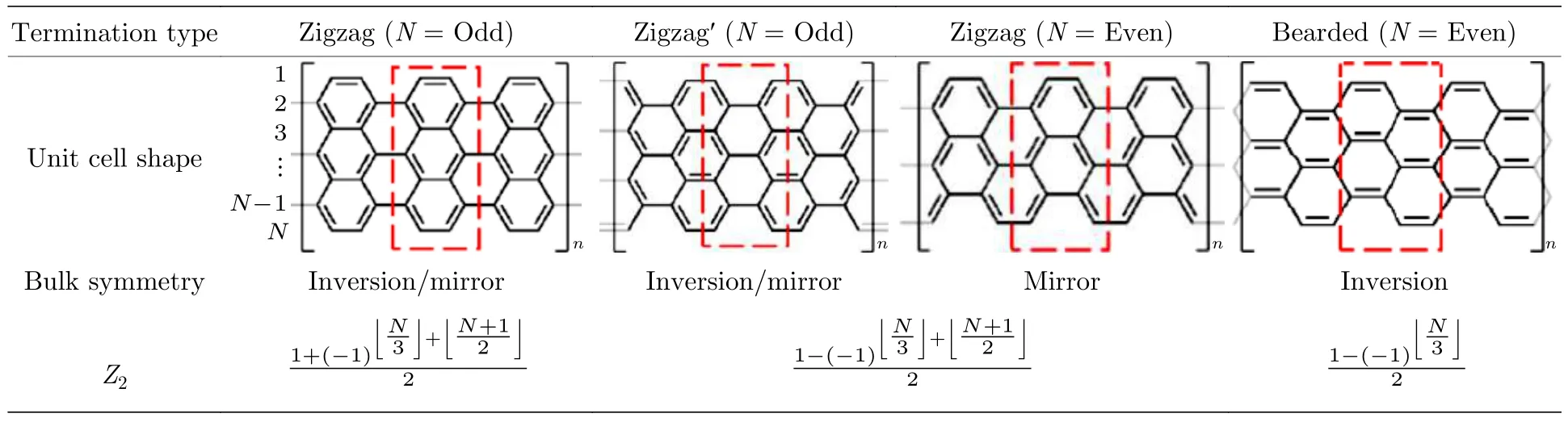
表1 AGNRs 的電子拓撲分類[121]Table 1.Categorization of electronic topology of AGNRs[121].
Gr?ning 等[122]在單晶表面上用小分子前驅體“自下而上”合成了所設計的石墨烯納米帶,并且通過理論模擬得出m≤ 3 時Z2=1,該模型構筑的石墨烯納米帶是拓撲非平庸的(圖10(b)).在實驗中利用小分子前驅體合成直型的具有擴展邊緣異質結構超晶格的石墨烯納米帶,超晶格是由耦合的鋸齒狀小片段組成,并且利用STS 技術結合理論模擬證明了所構筑出來m=3 的石墨烯納米帶,在其導帶和價帶之間產生了拓撲邊緣態(圖10(c)).幾乎在同時,Rizzo 等[123]預測出7—9 AGNR 超晶格將會在價帶和導帶間產生2 個能帶,分別為占據的拓撲誘導能帶和未占據的拓撲誘導能帶(圖11(b)).隨后,他們利用了相同的合成原理,“自下而上”合成出了事先設計好的石墨烯納米帶,圖11(a)是其化學結構模型以及不同結構的拓撲指數和高分辨率的STM 圖像.這些石墨烯納米帶由拓撲不變數變化的節點周期性排列構成,從而導致具有不同拓撲不變數的石墨烯納米片段異質結界面處拓撲態的形成.利用STS 譜學特征和理論計算模擬相結合(圖11(c)),可以證明不同石墨烯分子結構異質結處的拓撲界面態以及石墨烯分子結構拓撲邊緣態的存在[123].
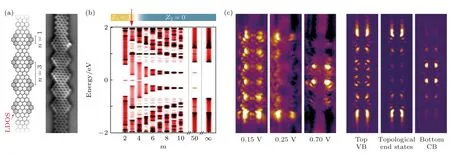
圖10 直線型邊緣擴展的AGNR 異質結構超晶格中的拓撲態 (a)化學結構和nc-AFM 圖像;(b)計算的能帶結構;(c)直線型邊緣擴展 AGNR 異質結構超晶格的局域態密度(LDOS)圖 [122]Fig.10.Topological states in in-line edge-extended AGNR heterostructure superlattices:(a) The chemical structure and nc-AFM image;(b) the calculated band structure;(c) the local density of states (LDOS) maps of an in-line edge-extended AGNR heterostructure superlattice[122].

圖11 7—9 AGNR 超晶格的拓撲態 (a) 化學結構模型以及不同結構的拓撲指數和高分辨率的STM 圖像;(b) 計算的石墨烯納米帶能帶結構;(c) 7—9 AGNR 超晶格的理論計算LDOS 圖以及對應的實驗dI/dV 圖[123]Fig.11.Topological states in a 7—9 AGNR superlattice:(a) The chemical structure and high-resolution STM image;(b) the calculated band structure;(c) the LDOS maps and corresponding experimental dI/dV diagram of a 7—9 AGNR superlattice[123].
5.2 石墨烯分子結構的自旋量子態測量和調控
設計合成特定的石墨烯分子結構,還可以引入局域自旋,實現無過渡金屬的僅由碳電子導致的磁性.磁性的引入可以通過設計特定形狀和邊緣結構,使其在π 電子的庫侖斥力作用下自發形成磁性;或者通過局部引入五元環打破石墨烯分子結構的二分圖對稱性;或者通過引入像三角烯這樣的自由基單元形成局域自旋.石墨烯分子結構中的自旋態具有顯著的優越性.首先,通過合理地設計邊緣或局域的化學鍵結構,可以精確地設計和調控石墨烯分子結構的磁性以及單自旋出現的空間位置,還可以設計多個自旋中心,并通過距離調控它們之間的相互耦合.其次,由于石墨烯分子結構全部由輕的碳元素組成,其自旋-軌道耦合和超精細相互作用極弱,有望具有較長的自旋態壽命和自旋擴散長度.
Li 等[124]通過設計小分子前驅體合成了具有3種不同異質結構拐點的石墨烯分子結構,如圖12(a)所示,結合掃描隧道顯微鏡(STM)和掃描隧道顯微譜(STS)技術,在1 型和2 型石墨烯分子結構中探測到了近藤效應(圖12(b)和圖12(c)),這表明在這些結構的拐角附近存在局域自旋.1 型結在PC 位置顯示近藤效應,因為它有1 個H 原子結合到ZZ 位置,使磁矩猝滅,而2 型結則相反.3 型結在2 個位置是都無額外的H 原子飽和,因此S=0 整體不顯示磁性,dI/dV譜上看不到近藤效應(圖12(d)).此外實驗中還使用了電子誘導脫氫讓1 和2 型轉變成3 型來調控石墨烯分子結構的磁性(圖12(e)—(h)).該工作證明了石墨烯納米條帶拐角結構的π 電子磁性,并且深入揭示了與自由基位點結合的額外氫原子會淬滅它們的磁矩,從而改變所制備石墨烯分子結構的自旋態.

圖12 (a) 3 種石墨烯納米結的DFT 理論模擬.(b),(c) 分別為類型 1 和類型 2 結上明亮區域的近藤共振.零偏置峰值主要在類型 1 結的4 個 PC 環和類型 2 結的3 個 ZZ 環上被檢測到.(d) 類型 3 結上零偏壓附近的雙峰特征.(e) 具有額外 H 原子2 個結的STM 圖.(f) 在電子誘導去除額外的 H 原子后STM 圖.(g),(h) 在脫氫過程之前(黑色)和之后(藍色)的 PC1 和 ZZ2 區域的dI/dV 譜.(g)中的插圖顯示了脫氫過程中的電流變化[124]Fig.12.(a) DFT theoretical simulation of three graphene nanojunctions.(b),(c) Kondo resonances over the bright regions of Type 1 and Type 2 junctions,respectively.The zero-bias peaks are mostly detected over four PC rings of Type 1 junctions and over three ZZ rings of Type 2 junctions.(d) Double-peak features around zero bias over Type 3 junctions.(e) STM image of two junctions with extra H atoms.(f) STM image after the removal of extra H atoms induced by electrons.(g),(h) The PC1 and ZZ2 regions of the dI/dV spectrum before (black) and after (blue) the dehydrogenation process.The inset in (g) shows the current during the dehydrogenation process [124].
利用相似的合成方法,Mishra 等[125]在超高真空條件下Au(111)表面上用三角烯作為基本結構單元制備出了一維自旋鏈,其中三角烯是一種自旋為S=1 的二自由基多環芳烴.三角烯中的磁性源于其二分蜂窩晶格中固有的亞晶格不平衡而產生的,這使得其具有類似自由基的凈自旋.三角烯及其衍生物難以通過傳統溶液化學路線合成,但最近已在一系列金屬和絕緣體表面上實現了原位合成,且其磁性可以在相對惰性的Au(111)表面上得以保持.因此,三角烯構成的自旋鏈(TSCs)為探索整數自旋S=1 鏈的拓撲物理提供一個理想的平臺.在該實驗中,通過使用STM 和STS 在原子尺度上探測了開放自旋鏈和閉合自旋環中與長度相關的磁激發,并直接觀察了其中的自旋激發能隙和分數邊緣態,如圖13(a)和圖13(b)所示為N=16 的開放三角烯鏈和閉合三角烯環的STM 圖像,圖13(c)為其上的dI/dV譜,可以看出只在開放三角烯鏈末端可觀測到近藤效應的信號,表明其邊緣態為S=1/2 的自旋態.圖13(d)為理論計算給出的自旋鏈模型.計算展示了N=2—16 的開放三角烯鏈的自旋激發能,可以看出邊緣激發能隨N的增加呈指數下降(圖13(e)).這一實驗結果非常漂亮地驗證了Haldane 早期的猜想,這是一種對稱保護的一維拓撲鏈,其邊緣S=1/2 的自旋態為無能隙的近自由態,而體態則具有能隙,與邊緣導電、內部絕緣的拓撲絕緣體具有類似的拓撲物性.
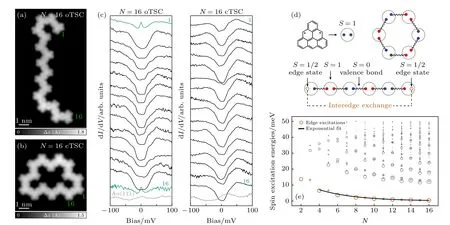
圖13 (a),(b) N=16 開放三角烯鏈(oTSC)和閉合三角烯環(cTSC) 的高分辨率 STM 圖;(c) 在N=16 oTSC 和cTSC 的每個單元上獲得的dI/dV 譜;(d) N=6 oTSC 和cTSC 的價鍵固態自旋態,占oTSC 中S=1/2 邊緣態,而cTSC 中沒有;(e) 對于N=2—16 的oTSC,由BLBQ 模型ED 計算的自旋激發能量[125]Fig.13.(a),(b) High-resolution STM images of N=16 oTSC (a) and cTSC (b);(c) dI/dV spectra acquired on every unit of the N=16 oTSC (a) and cTSC (b);(d) the valence bond solid spin state for N=6 oTSC and cTSC,accounting for S=1/2 edge states in the oTSC and their absence in the cTSC;(e) For oTSC with N=2—16,the spin excitation energy calculated from the ED of the BLBQ model[125].
在具有納米石墨烯結構的分子中引入五環結構,是實現全碳磁性的另一種手段.這主要是因為五元環的引入打破了正常石墨烯分子原本的二分圖對稱性,使得該單分子系統產生了自旋密度分布差和自旋極化.這樣的石墨烯分子結構稱為f-NG.一個典型的例子是Zheng 等[126]在金屬襯底上精確合成了f-NG 及其共價偶聯分子二聚體,并對其做了精細的STM 和STS 表征.在單個f-NG 分子上做dI/dV譜觀察到零偏壓處的近藤共振特征峰(圖14(a),插圖為f-NG 恒高的STM 圖,數字位置對應于做dI/dV譜位置).峰值強度與空間自旋密度強度成正比.由于不對稱的分子結構,共價偶聯的f-NG 二聚體具有6 種不同的構型(標記為C1—C6),這取決于2 個嵌入的五元環的位置.2 個五元環位于每個f-NG (c3 和c4 構型)的同一側,f-NG二聚體的基態為單線態,磁交換作用為3 meV.有趣的是,通過將2 個五元環相對放置(C5 和C6 構型),可以進一步增強交換相互作用,其中交換能量高達29 meV (圖14(b)),這使得設計出室溫的自旋電子器件成為可能.磁交換作用強度對嵌入五元環位置的強烈依賴性源于f-NG 單體內部不均勻的自旋密度分布.如圖14(c)所示,通過實驗研究了4 種不同的f-NG 二聚體,表示為C1—C4.對于C1,圖14(d)中的dI/dV譜在2 個f-NG 單元上都顯示出顯著的近藤效應,表明2 個自旋之間沒有強的磁耦合.對于C2 構型,五元環的位置與C1 相似,但2 個f-NG 單元相對于彼此傾斜,在2 個f-NG 長軸之間形成 133°的角.有趣的是,在C2 上dI/dV譜中觀察到費米面兩側對稱出現臺階,表明2 個自旋的反鐵磁耦合具有2 meV 的磁交換相互作用.對于C3 和C4 結構,dI/dV譜表明2 個自旋也是反鐵磁耦合的,但具有更大的磁交換能.這一實驗結果證實了單個f-NG分子具有S=1/2 的單自旋,而在共價連接的f-NG 二聚體中,2 個單自旋之間可實現反鐵磁耦合,其耦合強度隨二聚體的連接形式不同而有所不同,這為調控f-NG 石墨烯分子結構的自旋提供了思路.
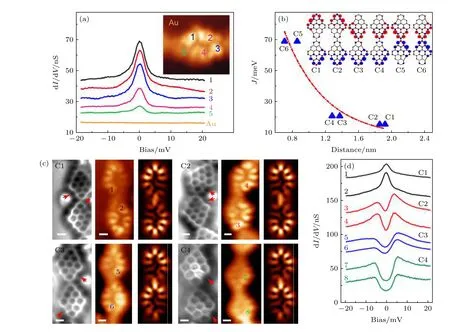
圖14 (a) 在插圖圖像中彩色數字標記位置做的 dI/dV 譜.(b) 磁交換作用是每個單元中自旋密度最大的2 個碳原子之間距離的函數.插圖為6 種不同f-NG 二聚體的自旋密度分布.所有二聚體都呈現單線態基態.藍色和紅色等表面表示自旋向上和自旋向下的密度.(c) 實驗觀察到4 種命名為 C1—C4 的 f-NG 二聚體構型.左側,nc-AFM 圖;中間,恒高的STM 圖,右側,模擬 STM圖.(d) 在 (c) 中標記的位置做的 dI/dV 譜[126].Fig.14.(a) dI/dV spectra taken at the positions marked by colored numbers in the inset current image.(b) The magnetic exchange interaction J as a function of the distance between two carbon atoms with the strongest spin density in each unit.Inset:spin density distribution of six different f-NG dimers.All dimers exhibit a singlet ground state.Blue and red isosurfaces denote spin up and spin down density.(c) Experimental observed four configurations of f-NG dimers named as C1—C4.Left,nc-AFM frequency shift image;middle,constant-height current image;right,simulated STM image.(d) dI/dV spectra taken at the positions marked in (c)[126].
6 總結與展望
本文綜述了基于STM 技術方向表面單分子量子態的制備、探測和調控的一些進展.我們可以看到,在技術方法上,STM 不僅可以實現對表面吸附單分子的空間高分辨和能量高分辨成像,還可以通過原位操縱以及結合外場和局域場等手段,實現對分子尺度的結構和電子態、自旋態等量子態的調控.除了直接操縱分子體系實現想要的人造分子結構,還可以通過STM 去操縱CO 分子構造出類似石墨烯的“反分子”人工晶格結構[127].STM 具備豐富的譜學方法,可以在表征單分子的隧道輸運性質的同時,利用dI/dV譜和圖像研究單分子體系局域態密度的空間分布,利用d2I/d2V二次微分譜,即非彈性電子隧穿譜(IETS),來研究單分子的振動激發和自旋激發.從STM 最早應用于單分子研究迄今,經過二十多年的發展,在STM 的基礎上已經結合不同的技術,發展出了眾多的聯合探測手段用于原位的量子態表征,例如:STM-ESR,STMgate[128,129],THz-STM,STML,STM-SQUID[130],STM-磁場[131]等.
在體系上,隨著表面原位合成技術的飛速發展和成熟,眾多不同結構石墨烯類分子的設計、合成和制備都成為現實.人工設計定制的這些石墨烯納米分子結構極大地豐富了表面單分子量子態的研究體系,其中拓撲電子態、局域自旋態的探測和表征已經展示出這一方向的豐富物理內涵.
在應用前景上,單分子發光對于構造單光子源以及信息傳輸有著重大的意義.分子天然具備有全同性,其發光頻率易于調控且均一性好,并且分子可以通過修飾基團來提供極為豐富的調節手段.但現階段半導體量子點的發光效率仍然更高并且制備更容易,單分子雖然有其優勢,但難以封裝,易受到外界干擾和影響等問題目前還沒有較好的解決方案.單分子作為可實用化的量子光源還需要等待微納技術的進一步發展.而表面單分子體系的拓撲物性、局域自旋等研究目前大多仍處于靜態探測的階段,要利用單分子體系的拓撲界面態、自旋態作為量子信息的載體,必須要解決來自襯底、分子環境等各方面對分子體系量子態相干性的破壞.而STM 作為一種近接觸式的探測和表征手段,如何在探測自旋量子態的同時避免和消除這些退相干因素,有待新的方案的提出,和ESR 的結合能夠部分解決探測的技術問題,但分子本身的隔離封裝依然沒有好的解決方案,目前使用的方法是在分子和金屬襯底之間引入絕緣層,但這樣隔離的分子難以在較高溫度下穩定吸附.最近對二維材料的研究或許可以帶來一些啟發.在二維材料的器件中,為了最大程度減少對二維材料性質的影響,常用的絕緣層和封裝層不再是硅基半導體常用的氧化物,而是使用單層六角氮化硼作為隔絕層.氮化硼非磁性且絕緣性極好,化學性質為惰性,與二維材料幾乎沒有電子和自旋相互作用.同樣地,如果能夠用氮化硼作為封裝隔絕材料,把單分子量子體系封裝在兩層氮化硼中,那么單分子的電子學性質、自旋性質、光學性質的量子特性是否有望像金剛石中的NV 色心一樣得到很好的保持,這需要實驗和理論的檢驗.我們期待這一領域下一階段重要的突破來自于分子體系量子態的有效封裝,可以使得單分子相干量子態的研究成為可能,從而實現對單分子體系優越量子特性的真正利用,為基于單分子量子態的量子信息提供物質載體.
猜你喜歡
中學化學(2024年5期)2024-07-08 09:24:57
中學化學(2024年2期)2024-06-17 04:01:47
小獼猴智力畫刊(2021年11期)2021-11-28 13:10:02
中學生數理化(高中版.高考理化)(2019年6期)2019-06-22 09:55:44
鉆井液與完井液(2018年5期)2018-02-13 01:07:44
中學化學(2016年10期)2017-01-07 08:37:06
新鄉學院學報(2016年6期)2016-12-01 05:21:37
湖南城市學院學報(自然科學版)(2016年2期)2016-12-01 04:06:40
化學教學(2015年6期)2015-09-15 02:50:02
中國石油大學學報(自然科學版)(2013年6期)2013-03-11 18:35:20
