HPLC同時檢測紅茶中兒茶素和茶黃素含量
2022-03-30 12:10:38費璠張梓瑩胡松徐丹紅劉仲華
食品與發酵工業 2022年5期
費璠,張梓瑩,胡松,徐丹紅,劉仲華,4*
1(湖南大眾傳媒職業技術學院,湖南 長沙,410100)2(湖南農業大學,茶學教育部重點實驗室,湖南 長沙,410128)3(國家植物功能成分利用工程技術研究中心,湖南 長沙,410128)4(植物功能成分利用省部共建協同創新中心,湖南 長沙,410128)
紅茶,作為當今世界最受歡迎的茶葉,占茶葉消費市場份額的70%~80%[1-2]。紅茶屬全發酵茶,主要分為工夫紅茶、小種紅茶、紅碎茶3大類[3-4]。其呈味物質和功能性成分包括兒茶素、茶黃素、茶紅素等,這些內含成分不僅滋味爽口,還兼具多種保健作用,如消炎、抗氧化、抗腫瘤、延緩衰老、保肝護肝等[5-8]。紅茶通過萎凋揉捻等工藝使得茶葉破碎,茶葉內含物與內源酶接觸,發生酶促氧化反應[9]。茶葉中大分子物質發生水解反應,兒茶素等多酚類化合物發生氧化聚合,形成大量香氣化合物及茶黃素、茶紅素、茶褐素等有色物質,構成紅茶鮮香馥郁、紅湯紅葉、滋味濃醇、回味悠長的品質特征[10]。兒茶素和茶黃素作為紅茶的重要品質因子,其含量高低直接影響著紅茶品質的優劣[11]。開發能快速準確的分析測定紅茶中兒茶素和茶黃素含量的方法對有效判斷紅茶品質、控制紅茶質量、指導紅茶的科學生產、實現高品質紅茶加工具有重要意義。
近年來,茶黃素和兒茶素檢測主要采用HPLC-MS和UPLC等檢測[12-13]。HPLC-MS和UPLC法設備采購價格較昂貴,儀器操作復雜,不適用于一般的實驗室條件。而HPLC-UV法是一種常見的快速檢測方法,設備價格低廉、易得,且分離效率高、準確度和精密度較好[14]。本研究基于HPLC-UV,通過優化檢測條件,建立了一種可同時測定兒茶素和茶黃素的高效液相色譜方法。
1 材料與方法
1.1 材料與試劑
乙腈、甲酸(均為色譜純),美國TEDIA公司;甲醇(分析純),國藥集團化學試劑有限公司;標準品咖啡堿(caffeine,Caf)、表沒食子酸酯兒茶素(epigallocatechin,EGC)、兒茶素(DL-catechin,DL-C)、表兒茶素(epicatechin,EC)、表沒食子兒茶素沒食子酸酯(epigallocatechin gallate,EGCG)、沒食子兒茶素沒食子酸酯(gallocatechin gallate,GCG)、表兒茶素沒食子酸酯(epicatechin gallate,ECG),成都普瑞法科技開發有限公司(純度≥98%);茶黃素(theaflavin,TF)、茶黃素-3-沒食子酸酯(theaflavin-3-gallate,TF-3-G)、茶黃素-3′-沒食子酸酯(theaflavin-3′-gallate,TF-3′-G)、茶黃素-3,3′-雙沒食子酸酯(theaflavin-3,3′-digallate,TFDG),美國Sigma公司(純度≥98%)。
6種紅茶樣品包含黃金茶紅茶、祁門工夫、祁紅毛峰、滇紅、一炮紅(臺灣)、坦洋工夫(粉碎后過40目篩,備用),湖南省茶業集團股份有限公司。
1.2 儀器與設備
1260高效液相色譜儀、安捷倫SB-C18色譜柱(4.6 mm×250 mm,5 μm),美國安捷倫公司;MS204TS電子天平,梅特勒-托利多國際貿易(上海)有限公司;SB-3200DT超聲波清洗機,寧波新芝生物科技有限公司;HHS-21-6電熱恒溫水浴鍋,上海精宏實驗設備有限公司;分析型超純水機,上海葉拓科技有限公司;JGY-2500研磨器,浙江省永康市敏業工貿有限公司。
1.3 樣品檢測
1.3.1 標準品溶液配制
分別精密稱取Caf、EGC、DL-C、EC、EGCG、GCG、ECG、TF、TF-3-G、TF-3′-G和TFDG對照品各12.5 mg(精確到0.000 1 g)于50 mL棕色容量瓶中,用體積分數為10%乙腈溶液溶解并定容,搖勻,得250 μg/mL的混合內含物質標準液。過0.45 μm微孔濾膜,進樣量為20 μL,調節方法參數進行HPLC方法優化。
1.3.2 HPLC檢測方法及優化
色譜柱:安捷倫SB-C18液相色譜柱(4.6 mm×250 mm,5 μm)。流動相A為0.1%(質量分數)甲酸-乙腈,流動相B為0.1%(質量分數)甲酸-水,流速1.0 mL/min,進樣量為20 μL,檢測波長為280 nm,柱溫30 ℃。梯度洗脫設置見表1。以上述條件作為最初條件,根據11種茶葉內含物質的保留時間、分離度等因素,對HPLC檢測方法的流動相酸度、柱溫及洗脫梯度等幾個方面進行優化。
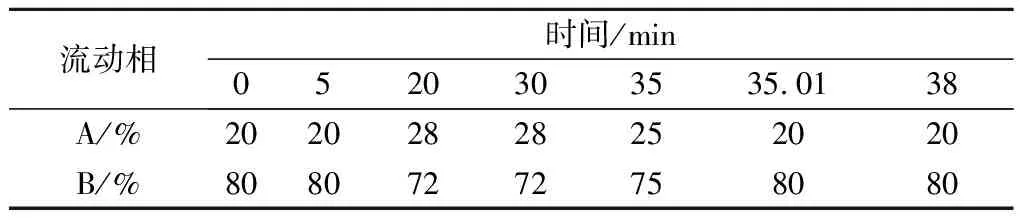
表1 高效液相色譜洗脫梯度
設置不同酸濃度A相0.05%、0.1%、0.15%、0.2%甲酸水溶液(體積分數),B相0.05%、0.1%、0.15%、0.2%甲酸乙腈溶液(體積分數)條件下11種茶葉內含物質分離情況。設置25、30、35 ℃ 3個柱溫對該方法分離效果的影響。根據峰型、分離度、保留時間等因素調整洗脫梯度以及檢測時間。
1.4 方法學驗證實驗
使用外標法定量分析檢測兒茶素和茶黃素的含量。分別精密稱取11種茶葉內含物質標準品各12.5 mg(精確到0.000 1 g)于25 mL棕色容量瓶中,用體積分數為10%乙腈溶液溶解并定容,搖勻,得500 μg/mL的混合內含物質母液。分別移取母液1、2、3、4、5、8 mL至10 mL棕色容量瓶中,用體積分數為10%乙腈溶液定容,得到50、100、150、200、250、400、500 μg/mL的標準工作液。各濃度標準品過0.45 μm微孔濾膜,進樣量為20 μL。以質量濃度(X)、峰面積(Y)繪制出標準工作曲線,并求回歸方程和相關系數。將最低濃度的標準溶液進樣,按照3倍信噪比(S/N)得到檢測方法的檢出限,按照10倍信噪比(S/N)得到檢測方法的定量限。
準確稱取使用研磨器均勻磨碎的滇紅茶樣品0.3 g于10 mL離心管中,加入5 mL體積分數為70%的甲醇溶液(70 ℃預熱),搖勻,于70 ℃水浴振蕩浸提10 min,冷卻至室溫,3 500 r/min離心10 min,取上清液于10 mL棕色容量瓶中。殘渣再重復上述步驟。合并提取液,定容至10 mL,搖勻,過0.45 μm濾膜,制得滇紅茶待測樣品。
以該方法檢測滇紅茶待測樣品,樣品重復進樣6次。根據所得峰面積分別計算11種茶葉內含成分的相對標準偏差(relative standard deviation,RSD),驗證該方法的精密度,重復性。
在待測滇紅茶樣品中分別加入一定量的11種茶葉內含成分對照品,進行加標回收率試驗,樣品重復進樣6次,考察該方法的準確度。加標回收率計算如公式(1)所示:

(1)
將滇紅茶待測樣液放置不同時間(0、2、4、6、8、12、24 h),以該方法檢測,根據11種茶葉內含成分的峰面積計算RSD值,考察方法的穩定性。
1.5 檢測紅茶樣品
分別準確稱取使用研磨器均勻磨碎的6種紅茶樣品0.3 g于10 mL離心管中,加入5 mL體積分數為70%的甲醇溶液(70 ℃預熱),搖勻,于70 ℃水浴中振蕩浸提10 min,冷卻至室溫,3 500 r/min離心10 min,取上清液于10 mL棕色容量瓶中。殘渣再重復上述步驟,合并提取液,定容至10 mL,搖勻,過0.45 μm 濾膜,制得紅茶待測樣品。使用上述優化后的方法檢測含有11種茶葉內含物質的對照樣,6種紅茶待測樣。每個樣品重復3次,外標法分析計算各茶樣11種茶葉內含物質的含量。
1.6 數據處理
所有試驗至少重復3次以上,使用Graphpad prism 8.0軟件進行圖表的繪制和相關數據的處理。
2 結果與分析
2.1 色譜條件的優化
乙腈-乙酸流動相體系中色譜圖峰形差、拖尾嚴重,本研究選擇甲酸調節酸濃度,設置流速1 mL/min,柱溫30 ℃,以A相甲酸乙腈溶液、B相甲酸水溶液為流動相,用甲酸調節酸濃度,考察不同酸體積分數A相0.05%、0.1%、0.15%、0.2%甲酸水溶液,B相0.05%、0.1%、0.15%、0.2%甲酸乙腈溶液條件下11種茶葉內含物質分離情況。結果發現,流動相A和B酸濃度相互搭配,酸濃度越低,待測物質保留時間越短,分離效果越差,出現多峰重疊現象;隨著酸濃度升高,11種茶葉內含物質分離效果越好,但保留時間也隨之增長,且酸濃度過高易對色譜柱造成影響。綜合考慮,流動相選擇A相甲酸體積分數為0.1%、B相甲酸體積分數為0.1%時,分離效果好,保留時間適中且對儀器損傷較小。
以A相含0.1%(體積分數)甲酸-乙腈溶液、B相含0.1%(體積分數)甲酸-水溶液為流動相,在進樣量為20 μL,流速為1 mL/min的條件下,分析25、30、35 ℃ 3個柱溫對該方法分離效果的影響。結果發現,隨著柱溫升高,11種待檢物質保留時間縮短,但分離效果變差,綜合分析,30 ℃時待測物質分離效果最佳。
洗脫梯度是影響色譜出峰順序和分離度的重要因素。本研究優化流動相洗脫梯度,在洗脫前期以8%的A相,92%的B相洗脫,能夠使兒茶素各組分完全分開且分離度高,峰型良好。待兒茶素組分全部洗脫后加大A相比例,茶黃素組分也均勻分開。經多次優化后,以表2條件進行梯度洗脫可有效分離兒茶素和茶黃素,11種茶葉內含物峰型完整,分離效果優異(圖1)。
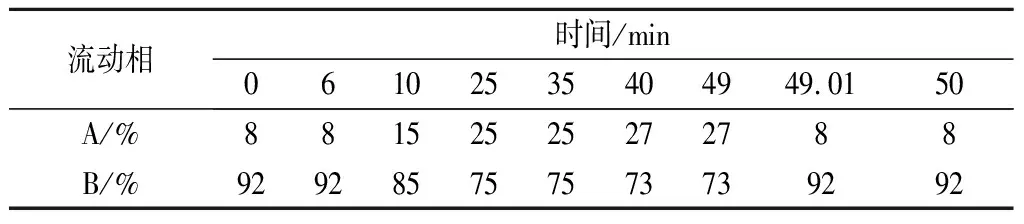
表2 高效液相色譜洗脫梯度
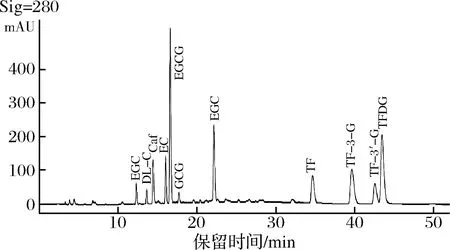
圖1 最優條件下11種內含物質標樣HPLC色譜圖
2.2 方法學驗證
檢測質量濃度50~500 μg/mL的一系列標準工作液,得到11條標準曲線。如表3所示,各物質標準曲線的R2均>0.999 0,說明各單體的質量濃度和色譜峰面積具有良好的線性相關性。11種茶葉內含物的檢出限范圍為0.21~11.13 μg/mL,定量限范圍為0.70~37.10 μg/mL。
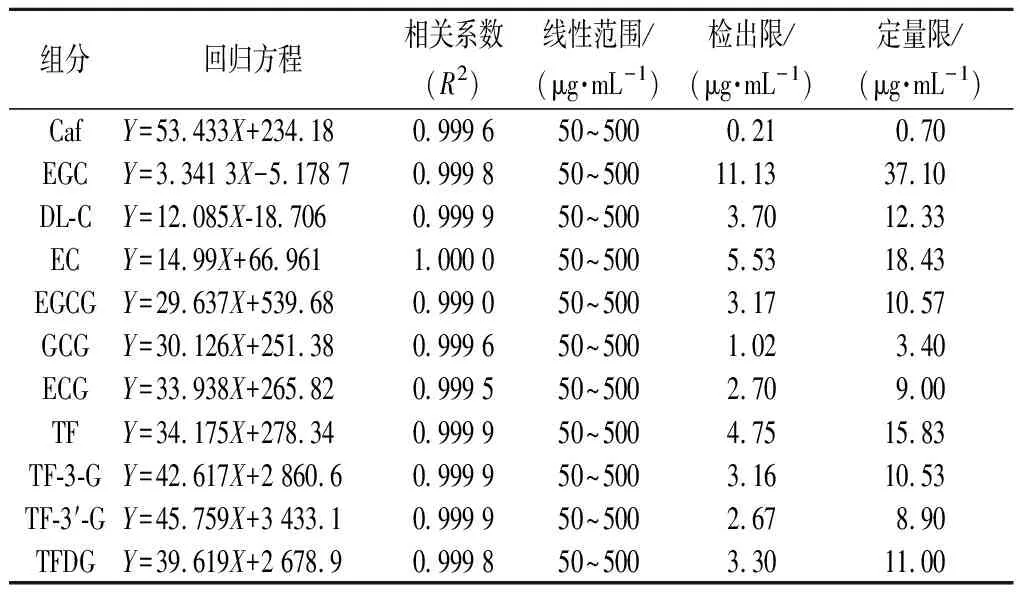
表3 11種茶葉內含物質的標準曲線
精密度、重復性、穩定性是衡量HPLC檢測方法準確性的一個重要指標。基于上述優化后的檢測方法,精密度、重復性和穩定性范圍分別為0.36%~3.40%、2.04%~4.82%、0.51%~3.83%(圖2),均<5%。
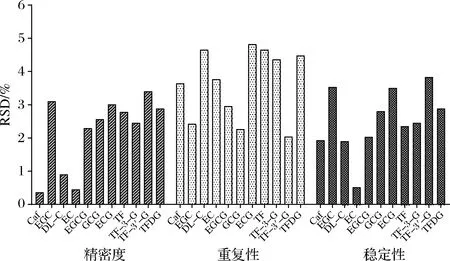
圖2 檢測方法儀器精密性、重復性、穩定性驗證
表明該方法具有良好的精密性、重復性、穩定性,是一種可穩定準確檢測茶葉中11種茶葉內含物質的方法。紅茶樣品加標回收實驗結果顯示加標回收率介于98.69%~104.77%,RSD均<5%(表4)。由此說明該方法準確度高,操作過程中損失少,回收率高。
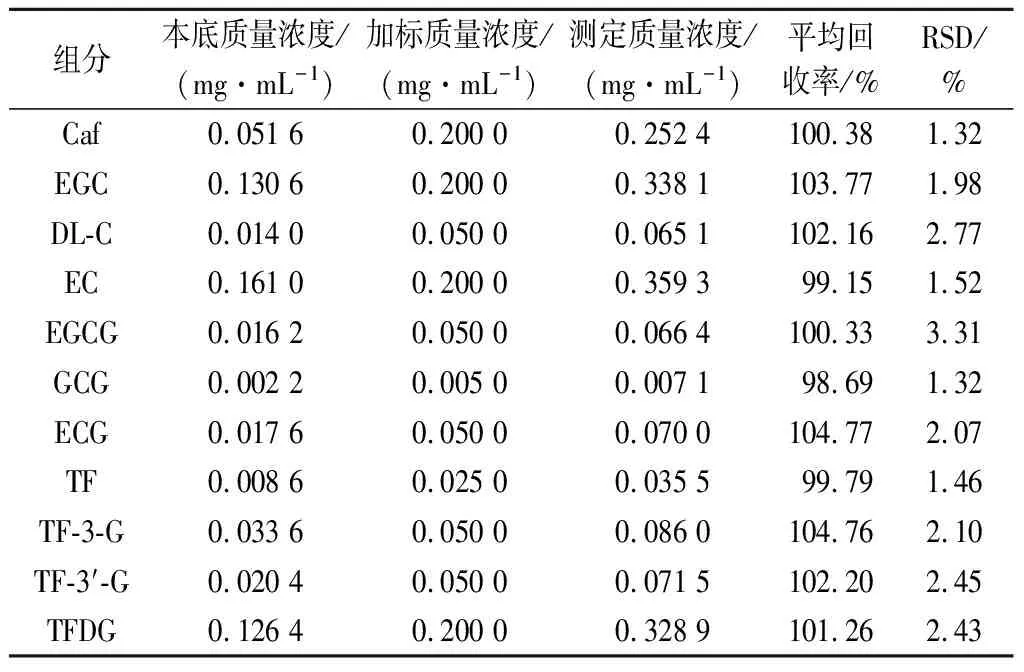
表4 準確度試驗結果(n=6)
2.3 紅茶樣品測定
基于上述優化后的檢測方法測定6種紅茶樣品,樣品中茶黃素總量從高到低依次為黃金茶紅茶、祁門工夫、祁紅毛峰、滇紅、坦洋功夫、一炮紅(圖3)。
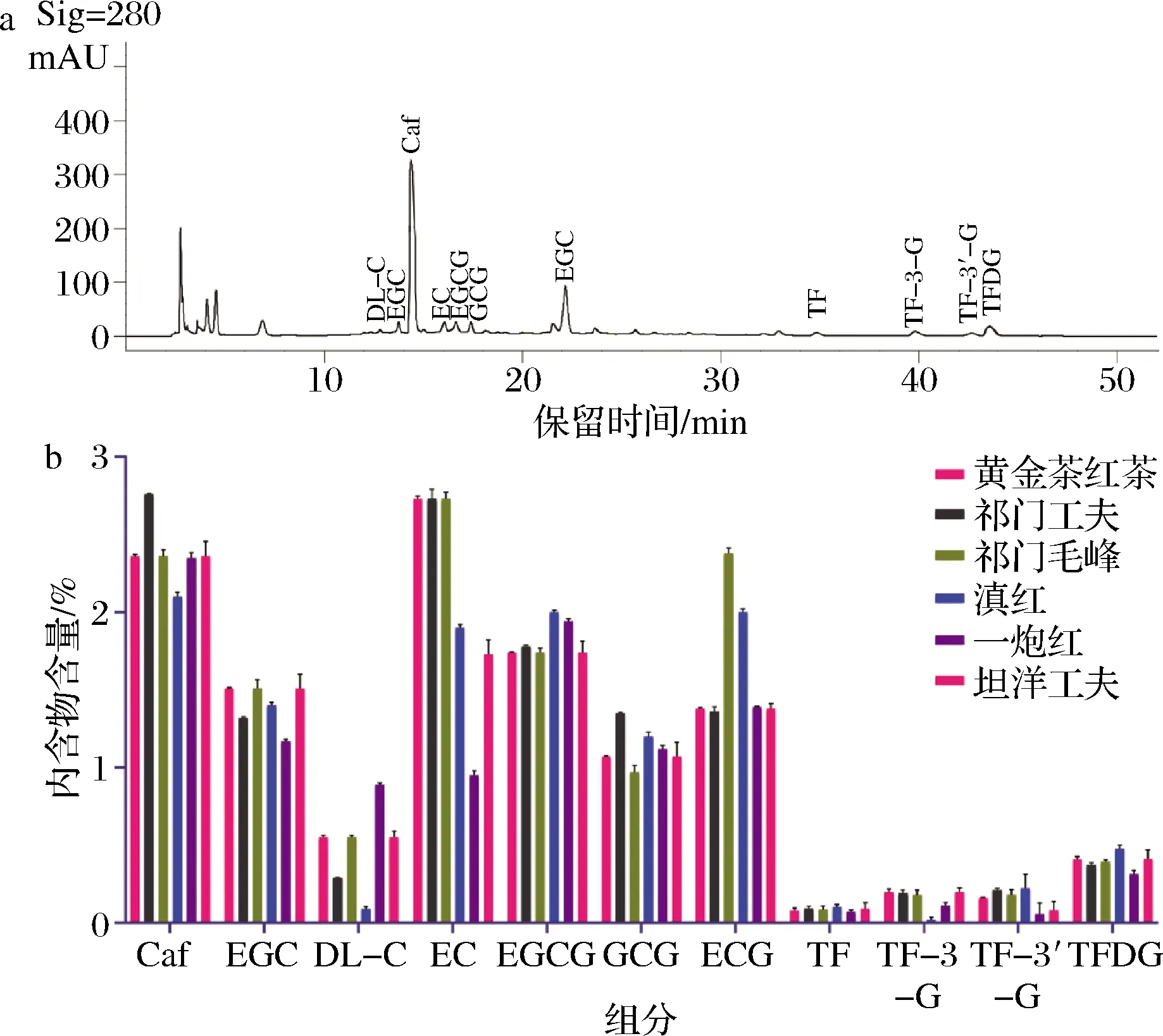
a-滇紅茶HPLC色譜圖;b-6種紅茶樣品中茶葉內含物含量
各樣品中4種主要茶黃素的含量范圍為0.554%~0.863%,其中TFDG在所有茶中含量最高。兒茶素方面,紅茶樣品均表現為簡單兒茶素的含量高于酯型兒茶素,這可能與茶鮮葉中的酯型兒茶素在發酵過程中被水解為簡單兒茶素,且酯型兒茶素在酶的催化作用下氧化聚合成茶黃素、茶紅素等有關,由此造成紅茶中的酯型兒茶素含量較低。各樣品中咖啡堿含量最高為2.76%(祁門毛峰),最低為2.1%(滇紅茶)。所有檢測值均在標準曲線有效測定范圍內。
3 結論
HPLC法是利用高壓泵將流動相壓入色譜柱內,待測組分經過分配、吸附或者離子交換過程來完成分離,最后經過檢測器進行檢測的一項分離技術[15-16]。HPLC具有分離度好和準確度高的特點,是目前檢測兒茶素應用最廣泛、技術最成熟的方法。劉錦文等[17]以V(甲醇)∶V(1%磷酸水溶液)=68.32為流動相,通過Diamonsil C18色譜柱分離,在279 nm條件下檢測到ECG和EGCG且分離效果好。付靜等[18]利用UPLC檢測出了綠茶中的5種兒茶素(C、EC、EGC、ECG、EGCG)均能達到較好的基線分離效果,方法具有良好的重復性和準確性。張爽等[19]建立了一種超高效液相色譜-紫外檢測器測定茶飲料中沒食子酸(gallic acid,GA)和5種兒茶素(C,EC,EGC,ECG,EGCG)的方法,6種組分在7 min內達到完全分離且各組分線性范圍良好。盡管UPLC已運用于茶黃素、兒茶素的檢測中,但儀器較昂貴,普及度不高,實驗操作要求嚴格,難以大規模推廣,且現有的檢測方法不能同時測定兒茶素和茶黃素含量,或是存在分離度差,定量不準確等缺陷[20-21]。
本研究在HPLC檢測茶黃素的基礎上優化檢測方法,建立了一種使用HPLC可同時、快速、準確、高效檢測紅茶中Caf、EGC、DL-C、EC、EGCG、GCG、ECG、TF、TF-3-G、TF-3′-G和TFDG等11種物質含量的方法。經流動相酸濃度、流速和柱溫的優化,確定了最佳色譜條件。同時考察了線性范圍、相關系數,并進行精密度、重復性、穩定性和準確度試驗,RSD均<5%,加標回收率介于98.69%~104.77%,最后使用該方法對6種紅茶中的兒茶素和茶黃素含量進行了檢測和比較。本研究為紅茶加工和生化品質研究提供了一種快速可靠的檢測方法,以期更好地促進紅茶產業發展。
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產業(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12