高電壓鈷酸鋰電池的研究進展
2022-03-30 03:35:48程梅笑萬廣聰申海鵬郭營軍李新麗
電源技術 2022年3期
程梅笑,萬廣聰,申海鵬,郭營軍,2,李新麗,2
(1.湖州昆侖億恩科電池材料有限公司,浙江湖州 313103;2.香河昆侖化學制品有限公司,河北廊坊 065400)
鈷酸鋰(LCO)在所有鋰離子電池正極材料中具有體積比能量高,工作電壓范圍寬,壓實密度高,理論比容量大,且LCO 特殊的α-NaFe2層狀結構可以實現Li+的快速遷移及穩定循環;但是,LCO 材料的實際比容量(140 mAh/g,Li1-xCoO2,x≈0.5,~4.2 Vvs.Li/Li+)只有理論值(274 mAh/g,Li1-xCoO2,x≈0.5,~4.2 Vvs.Li/Li+)的60%[1]。研究表明,通過提高電池的充電截止電壓,可以大大提高LCO 正極材料的比容量以及能量密度,然而隨著Li+的不斷脫嵌,導致LCO 從六方晶相(O3相)到單斜晶相的不可逆相變[2-3]。此外,在高電壓下LCO 材料界面與電解質間的副反應通常會導致LCO 電池容量下降及循環性能不穩定,從而限制了高電壓LCO 電池的商業應用。為了充分發揮LCO 材料的應用價值,研究者進行了大量的研發工作,主要包括LCO 正極材料的改性及電解質添加劑的篩選。本文將著重介紹高電壓LCO 電池正極材料的改性方法、電解質的優化方案及其中涉及的機理分析。
1 正極材料的研究進展
在鋰離子電池充電到高截止電壓的過程中,LCO 晶體結構經歷了多種相變(H1 到H2,~3.9 V,絕緣體-金屬轉變;M1,~4.1 V;H3,~4.2 V,有序-無序轉變;M2,~4.55 V;O1),導致晶體向c和a軸各向異性膨脹和收縮[4-5]。反復經歷上述過程后,LCO 材料不可逆相變(例如,H2 到M1,M1 到H3,H3 到M2)增多,導致鋰離子電池的容量衰減嚴重[6]。
1.1 摻雜法
體相元素摻雜可以改變LCO 材料原子級晶格結構,例如調節晶格缺陷比例,重排陽離子,重新分布電荷和電子結構,是提高LCO 材料結構穩定性最廣泛使用的方法[7]。摻雜的元素分為過渡金屬(Cu,Ni,Mn,Ti,La 等)及非過渡金屬(Mg,Ca,Al,Si 等)兩大類[8-9]。在晶體結構中摻雜元素通過占據鋰離子或者鈷離子的空間位置,達到穩定脫嵌鋰晶體、擴大晶格間距及支撐層狀結構的效果,進而抑制正極材料鈷離子溶出及提升循環壽命[10]。
現階段,研究者通過多種元素的協同摻雜進一步提升LCO 的電化學性能。Zhang 等[11]通過Ti-Mg-Al 共摻雜實現了LCO 電池(vs.Li/Li+)在4.6 V 下的穩定循環。在鋰離子電池中,以0.5C倍率100 次循環后,仍實現了174 mAh/g 的高可逆放電比容量(與第二個循環相比,容量保持率86%)。研究表明,Mg 和Al 原子已成功摻雜到LCO 晶格中,從而改變了(去)鋰化過程中的相變行為。Mg 的摻雜還可以增加材料的電子電導率。相反,即使微量的Ti 也不能完全摻入LCO 晶格中。Ti 在晶界和表面的偏析,一方面改變了樣品顆粒的微觀結構,這有利于整個Li+的擴散和均勻的內部應變分布;另一方面,抑制了氧的活性并使其穩定在高充電電壓材料的表面。圖1為三元素摻雜的空間及數量分布。電極材料的完美設計不僅需要探究摻雜元素的種類,也需要探究摻雜元素的用量。Xu等[12]探究了多元素(Al,Ti 和Mg)摻雜LCO 材料中Mg 用量對LCO 電化學性能的影響,結果表明,過量的Mg 摻雜不利于LCO 材料內部離子的動力學。這是由于過量的Mg 在LCO 材料表面形成了約2 nm 的MgO 涂層以及在內部隨機形成MgO顆粒,導致Li+在晶體中脫嵌困難且鋰離子電池放電平臺降低。在綜合考慮改性材料的循環性能(25 ℃,0.1C,200 次,容量保持率96%)、比容量(190.2 mAh/g)和動態性能(0.2C、0.5C、1C、2C以及0.2C,每種倍率5 個循環,最終容量保持率95%)的基礎上,LCO 材料中摻雜0.12%(質量分數)的Mg 最適宜。Song 等[13]認為高電壓LCO 材料中的多元素摻雜通常會由于晶界處的元素偏析而引起多晶化,這些元素偏析可以實現相鄰初級粒子之間的穩固連接和緩沖,抑制了晶界處裂解和形成裂紋,并提供長期的穩定結構。
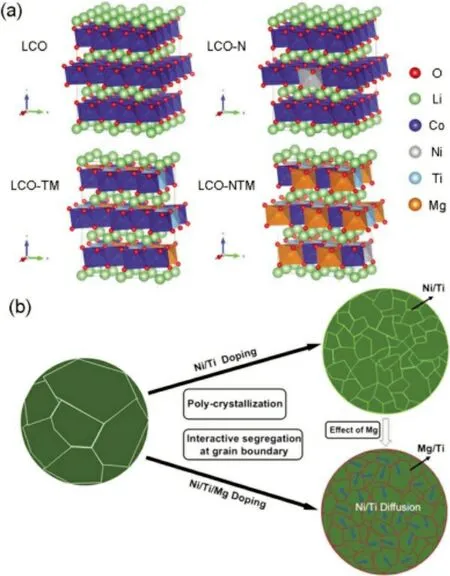
圖1 (a)摻雜元素之間相互作用的理論計算與(b)多種元素摻雜的影響[13]
摻雜后的LCO 材料也有不穩定因素存在:(1)摻雜元素與電解質接觸面積較大,導致嚴重的副反應;(2)摻雜元素各向異性膨脹,導致次級顆粒破裂;(3)摻雜元素在電解質中溶出。
1.2 包覆法
表面包覆法可以有效穩定LCO 材料界面,抑制不良反應進而提高高電壓下鋰離子電池的電化學性能[14]。表面包覆的材料通常具有高機械強度及化學穩定性,主要包括金屬氧化物(例如:Al2O3,CuO,Y2O3),金屬氟化物(例如:MgF2,CeF3,AlF3)和金屬磷酸鹽(例如:AlPO4,Li3PO4)等[15-16]。圖2 為LCO材料包覆方法及示例。這些包覆材料通過在LCO 表面形成兩相復合物(固溶體)而產生穩定的晶界,使去鋰化狀態下LCO 材料熱穩定性增強并隔絕與電解質之間的副反應;但是LCO 的可逆容量降低,循環時電荷轉移阻抗增加[17]。
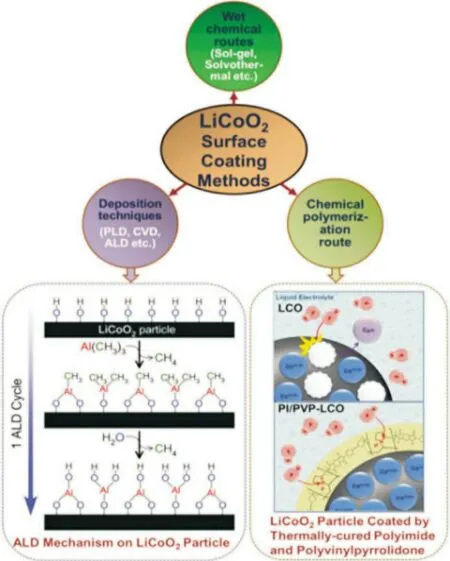
圖2 LiCoO2的表面包覆方法流程圖以及示例[17]
因此,研究者們為解決上述問題進行了實驗探究。Cheng 等[18]提供了一種具有多層結構的表面改性方法,即在LCO 外層包覆富鋅表面涂層,巖鹽相緩沖層和表面梯度Al 摻雜層,實現了LCO 電池在4.6 V 下的穩定循環,并且修飾后的涂層抑制了LCO 材料與電解質的界面反應及阻抗的增長。材料表面準外延層生長現象表明,多層結構顯著降低了LCO材料與表面涂層之間的晶格失配,增強了富鋅外層的穩定性。此外,無序的巖鹽相層和Al 表面摻雜也增強了結構穩定性。所有這些協同作用促使LCO 在4.6 V 下穩定循環,在500次循環后容量保持率為65.7%。一些研究人員認為,涂層可以抑制LCO 材料結構從H2 到M1 的相變,但是其他研究人員認為,包覆涂層不能抑制相變,但可以使LCO 材料晶相更具可逆性。Hu 等[19]利用濕化學法綜合元素摻雜和表面包覆的優點,對LCO 材料進行了Ba 和Ti 二元混合改性,處理后得到LiCoO2(LCO@BT),即LCO 表面上形成的穩定的固態Li-Ti-Co-O 薄膜,同時摻雜少量BaTiO3和TiO2顆粒。研究表明,改性層可促進表面Li+擴散,降低電荷轉移阻抗,并保護LCO 免受表面副反應引起的腐蝕。根據原位NMR 表征,在第一次電化學脫/嵌鋰之后,發現LxCO@BT(0.98<x<1)中晶體O3-I型可逆轉變,并且在4.5 V 的高截止電位下,改性材料初始放電比容量為190.5 mAh/g;0.2C,100 次循環后,容量保持率高達90.29%,放電比容量為180.4 mAh/g。
但是在實際工業生產過程中,受原料成本、制造環境及加工工藝等方面的影響,所面臨的問題則是均勻包覆LCO 材料表面。
2 電解質的研究進展
高電壓下,LCO 層狀結構由于過度脫鋰而變得不穩定,從而誘導Co4+溶解在液體電解質中;電解質在Co4+催化及高電勢雙重作用下被氧化分解生成大量氣體。此外,溶解的Co4+遷移到負極并沉積在表面,導致電解質進一步還原分解并消耗活性Li+。常規的電解質碳酸酯類溶劑在Co4+的催化作用下往往達不到理論氧化電勢(5 V,vs.Li/Li+)而在低電勢(≤4.5 V,vs.Li/Li+)下就被氧化分解[20]。因此,開發具有較高的氧化電位,同時又可以抑制LCO 表面Co4+溶出的電解質,是高電壓LCO 電池對電解質材料重要的要求。
2.1 電解質溶劑
理想狀態下,鋰離子電池負極的電化學勢需要低于電解質溶劑的最低未占據分子軌道(LUMO)能級,正極的電化學勢需要高于電解質溶劑的最高未占據分子軌道(HOMO)能級。因此高電壓鋰離子電池的進一步發展,需要電化學窗口大于5 V 的電解質溶劑。非對稱無環砜基電解質溶劑已成為具有高電化學穩定性(>5.0 V,vs.Li/Li+)的最新電解質溶劑之一。Wu 等[21]通過前線分子軌道理論計算發現乙基甲基砜(EMS)、四亞甲基砜(TMS)和乙基乙烯基砜(EVS)具有相對較好的氧化穩定性。這三種砜從高到低的氧化穩定性為EMS、TMS 和EVS,氧化分解電位都在5 V 以上。圖3 為鋰鹽和砜溶劑的電子云結構。除了砜類溶劑外,氟代溶劑也是一種良好的高電壓電解質溶劑,例如氟代線性碳酸酯,氟代環狀碳酸酯,氟代醚等[22]。氟原子具有很強的電負性和弱極性,氟取代氫會有效提高溶劑的氧化分解電壓,氟化后的溶劑通常比原始溶劑的抗氧化能力高,浸潤性好,符合高電壓電解質溶劑的要求[23]。不過氟化后的溶劑對鋰鹽的溶解度降低,在氟代溶劑中往往需要添加助溶劑。Yan 等[24]將乙酸乙酯氟化后制備出DFEAc,由于氟原子具有很強的吸電子能力,因此提高了EA 的氧化耐受能力,并且DFEAc 還繼承了EA 的低粘度,同時起到了高電壓溶劑及助溶劑的作用;具有新型電解質的鋰離子電池(vs.Li/ Li+)在4.6 V 下經過100 次循環后實現容量保持率89.23%。
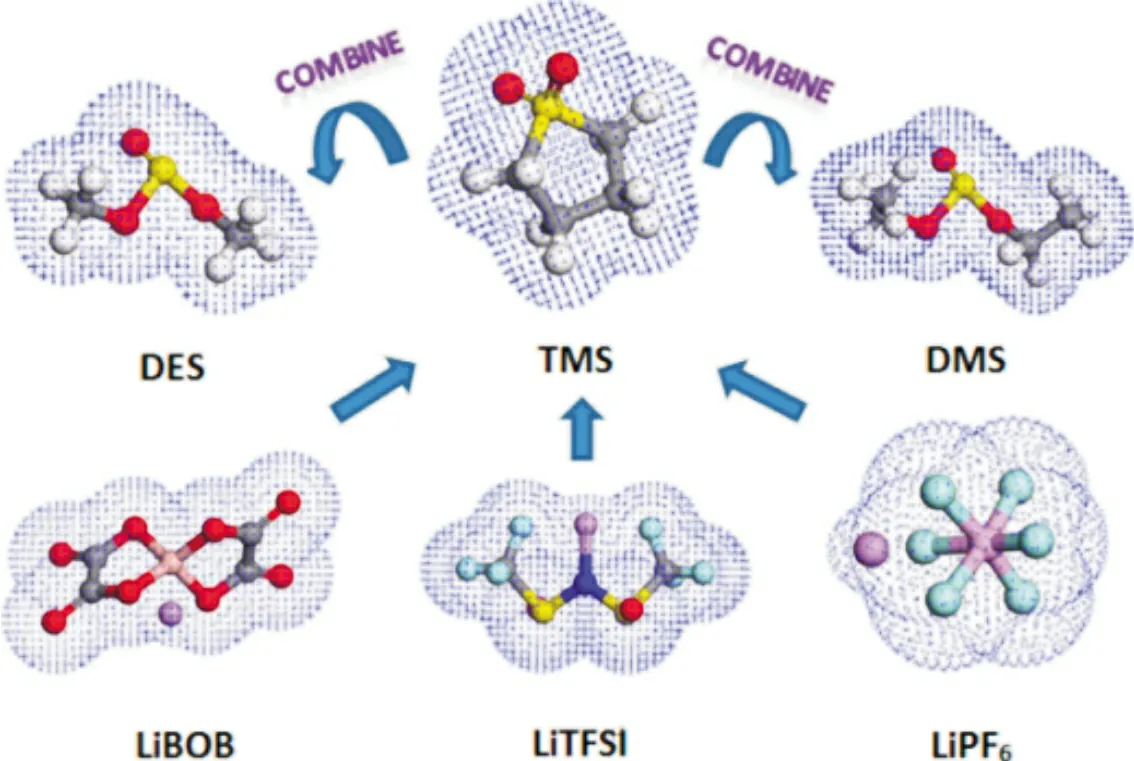
圖3 鋰鹽和砜類溶劑的電子云結構[21]
高電壓溶劑不僅有砜類溶劑和氟代溶劑還有腈類溶劑及離子液體等,但是他們各有優缺點,例如砜類溶劑及腈類溶劑粘度大,與負極兼容性差,氟代溶劑制作成本及安全要求高。為了趨利避害,研究者往往采用多種溶劑混合的方式來達到更優的效果,Kong 等[25]使用碳酸二甲酯/氟乙烯二丙醚/氟代碳酸乙烯酯/二甲基砜作為新型混合溶劑,與常規電解質相比,LCO 電池(vs.Li/Li+)在4.45 V 下循環300 次后,容量保持率從原來的38.1%提高到87.1%,在4.55 V 的更高電壓下300 次循環后容量保持率也從以前的21.0%有效地提高到74.9%,并且該電解質與石墨負極保持很好的相容性。
2.2 成膜添加劑
相較于在正極材料上摻雜和包覆無機化合物來抑制鈷離子溶出,在電解質中添加成膜添加劑是簡單有效且成本低廉的方法。根據前線分子軌道理論,正極成膜添加劑分子的氧化電位需低于電解質溶劑,HOMO 能級應高于溶劑分子。
研究發現,一些含S 的成膜添加劑分子HOMO 能量高于常見的碳酸酯溶劑,這表明它們的氧化電位較低,在充電過程中優先在正極上氧化聚合(形成Li2SO3或ROSO2Li),形成一層保護膜抑制鈷離子溶出及界面反應[26]。Zheng 等[27]在4.5 V的LCO 電池電解質(LiPF6,EC,DMC 和EMC)中加入0.5%二(甲基磺酰基)乙烷(DMSE)進行評估;在100 次充放電后,容量保持率從20.8%提高到66.5%。與碳酸酯溶劑相比,DMSE 具有更高的HOMO 能級和更低的氧化電位,由于較低的氧化穩定性,DMSE 會在LCO 表面上優先于溶劑分解形成CEI層,從而保護正極材料結構,抑制電解質溶劑進一步分解。在各種含S 添加劑中,噻吩類衍生物可以在電化學環境下聚合成聚噻吩,表現出高電導率和優異的化學穩定性。Sun 等[28]通過在基礎電解液中添加0.5% 2-(三氟乙酰基)噻吩(TFPN),LCO電池的循環(0.5C,3.0~4.4 V,100 次,vs.Li/ Li+)容量保持率從33.2%提升至90.6%。
與其他成膜添加劑不一樣的是腈類添加劑還可以與過渡金屬(Co4+或Ni4+)絡合,形成強配位鍵[R-CNδ-Metal(4+δ)+],防止過渡金屬對電解質催化氧化,從而改善電池的循環壽命及熱穩定性。Yang 等[29]通過在傳統碳酸酯類電解質中添加亞甲腈(SUN)或1,3,6-己三腈(HTCN)可以實現LCO 電池(vs.Li/Li+)在4.6 V 截止電壓下,300 次循環(30 ℃,1C)后容量保持率超過72%,200 次循環(55 ℃,1C)后容量保持率超過60%。這些改善得益于其在LCO 正極上形成超薄且均勻的CEI 膜,并抑制了微觀結構的演變(Co4+孤對電子對與-CN 的N 2p 軌道的配位),可以有效降低Co4+的高價態,從而降低了過渡金屬離子對整體電解質的催化反應性。
除此之外,還有含硼類添加劑[雙草酸硼酸鋰(LiBOB),草酸硼酸鋰(LiDFOB)和三(三甲基硅基)硼酸酯(TMSB)等]不僅可以優先在正極被氧化參與修飾CEI 層,還可防止PF6-分解為PF5、LiF 或其他副產物,親電子的含硼化合物也可以增加LiF 的溶解度,防止界面阻抗增加[30]。此外,硼酸鹽化合物可以被認為是陰離子受體,它們與鋰鹽中的陰離子絡合,不僅可以提高電解質中鋰離子的導電性,而且可以提高鋰離子的遷移數。
3 結論
摻雜元素的離子半徑比鈷離子大,因此它們可以在鋰離子嵌入/脫出過程中穩定層狀結構(立柱效應)。盡管對摻雜方法的研究已經進行了很長時間,但仍存在許多未知領域需要研究,例如F-的影響,多元素摻雜的影響以及抑制相變的機理等。包覆法可以構建LCO 材料保護層,以防止電解質對其腐蝕,并且與元素摻雜方法不同的是涂層使相變可逆而不是抑制相變。但是包覆法的難點在于涂層材料的鋰離子遷移率。就液體電解質而言,多采用前線分子軌道理論計算與電化學實驗相結合的方法探究其性能,電解質溶劑需要有更寬的HOMO 及LUMO“能帶隙”,才能保證在高電壓下溶劑整體性能的穩定,電解質添加劑則需要比溶劑高的HOMO 能級,以便在正極形成CEI。
