受損腺嘌呤與氨基酸殘基堆積復合物的ABEEM極化力場
2022-04-01 11:26:02鄭明杰
遼寧師范大學學報(自然科學版) 2022年1期
關鍵詞:體系
劉 翠, 王 洋, 鄭明杰, 尚 尉, 鄭 利
(遼寧師范大學 化學化工學院,遼寧 大連 116029)
細胞基因組常會因為水解和暴露于細胞內環境而導致正常堿基氧化和烷基化,發生突變,使DNA不能進行正常的復制和轉錄工作.損傷堿基數量積累過多將無法與生物體相容,除非它們被移除,否則會引發疾病,嚴重時會引發癌癥[1].此種內源性損傷必須通過DNA修復途徑中的堿基切除修復(BER)才能有效糾正,使基因組恢復正常.
堿基切除修復過程中涉及多種酶,其中用于特異性清除損傷堿基的酶稱為DNA糖基化酶[2],它去除受損堿基的作用機理是切割核苷酸的糖苷鍵.在這一過程中,蛋白質與堿基間除了氫鍵作用外,堆積作用也至關重要.堆積作用是一種重要的非鍵作用[3],它主要發生在π-π共軛體系中,對于增強體系的穩定性具有重大意義.π-π堆積作用的研究非常廣泛,包括了氨基酸與陽離子堆疊、糖-π堆疊、氨基酸-碳水化合物堆疊以及氨基酸-堿基的堆疊[4-6].例如Ebrahimi等人利用標準法和梯度平衡校正法優化尿嘧啶U與苯丙氨酸PHE、Na+和PHE之間的π相互作用;Gattani等人提出了一個平衡預測器——StackCBPred,基于從進化驅動的序列輪廓中提取的特征,預測蛋白質-碳水化合物結合位點.
堿基與芳香環氨基酸之間的π-π堆積作用近年來一直受到國內外研究學者的關注.例如:Baker等人[7]對氨基酸與堿基之間的相互作用進行研究,對比了蛋白質與核酸復合物的堆積作用的強弱,為修復酶識別堿基提供理論依據;Rutledge等人[8]通過掃描勢能面,研究了腺嘌呤、損傷腺嘌呤(3-甲基腺嘌呤)與氨基酸殘基之間的堆積作用,得出烷基化可增強修復酶活性部位的堆積相互作用,并給出了這種增強程度的度量.
目前為止,關于π-π堆積作用的研究包括化學實驗法和理論計算方法兩大類,而單純的實驗方法(如NMR和X射線晶體)雖然可以揭示DNA-蛋白質相互作用的結構,但是不能明確給出二體的相互作用強度信息.計算化學可以提供相關作用細節.Maria等人[9]在MP2/cc-pVDZ理論水平上,利用XMCQDPT2/sa2-CASSCF(12/12)方法對HBDI(4-羥基亞芐基-1,2-二甲基咪唑啉酮)與芳香族化合物之間的π-π堆積作用進行研究,并證明了變色位移與偶極矩的相關性.Stefan等人[10]利用ONIOM(QM:MM)方法,揭示了AAG(烷基腺嘌呤DNA糖基化酶)的活性位點結合不同的底物對脫糖基化的顯著影響.
量子力學方法計算精準,但計算的體系大小有限.對于復雜的生物大分子體系,必須要考慮內部復雜的環境、極化效應、電荷轉移效應、氫鍵作用及堆疊作用.常用力場,如AMBER[11]、OPLS[12]和AMOEBA[13]等,不能精準處理極化效應和電荷轉移效應.因此發展一種快速、準確的極化力場,模擬修復酶與損傷堿基的堆積作用十分必要.
Yang等人將基于密度泛函理論(DFT)和電負性均衡原理建立的ABEEM電荷模型與分子力場相結合,建立ABEEM極化力場[14].此力場通過多位點模型,及位點電荷受環境和結構變化而變化,體現生物分子中的極化效應和電荷轉移效應,而且計算速度快,適合于復雜生物大分子體系的模擬.
本工作通過對比從頭算的基準結果,調節π位點電荷和堆疊作用函數,發展了適用于堆疊作用的腺嘌呤和氨基酸殘基的ABEEM極化力場,在ABEEM極化力場中模擬4種受損腺嘌呤(次黃嘌呤(HYP)、1-N6-乙烯基腺嘌呤(εA)、和3,9-二甲基腺嘌呤(3,9MeA)、3-甲基腺嘌呤(3MeA))與4種芳香族氨基酸殘基(組氨酸(HIS)、色氨酸(TRP)、苯丙氨酸(PHE)、酪氨酸(TYR))的堆積作用.獲得有關DNA堿基與氨基酸殘基之間堆積作用幾何結構、電荷分布和作用強度的信息.由于這些相互作用在生命體系中扮演著無可替代的作用,因此該研究課題能為酶識別損傷腺嘌呤的機理提供理論指導.
1 模型分子
在研究氨基酸與堿基堆疊二聚體時,如果考慮蛋白質骨架或DNA骨架,會使得二聚體的相互作用強度發生變化,為了準確計算π-π堆積相互作用能,磷酸糖主鏈和氨基酸的蛋白質主鏈都用氫原子代替,各損傷腺嘌呤結構如圖1.
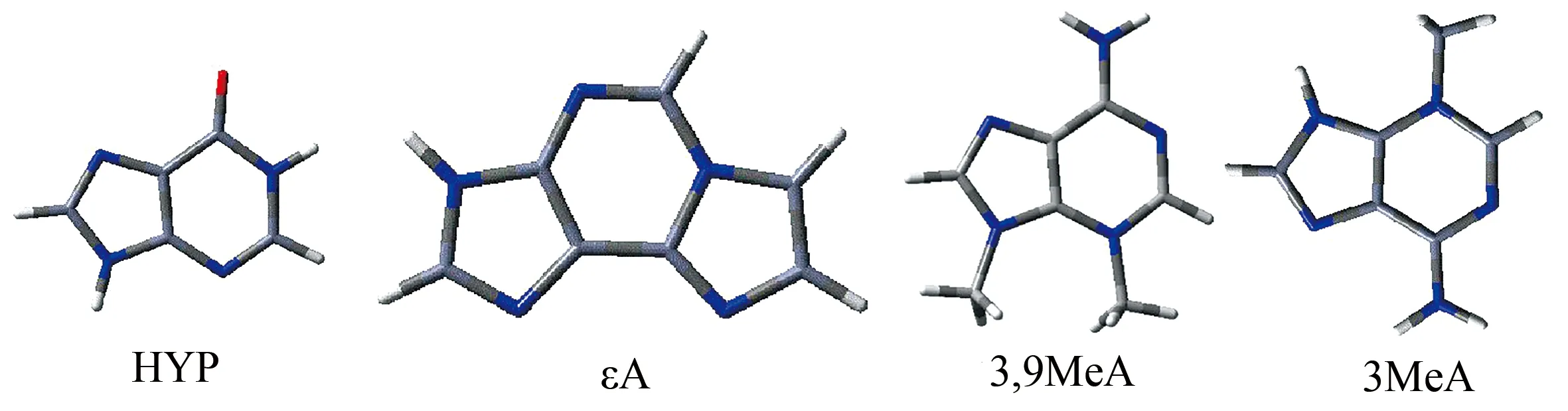
圖1 4種損傷腺嘌呤模型圖
氨基酸殘基與受損腺嘌呤之間的π-π堆積作用會受到4種因素的影響,分別是垂直位移R1、旋轉角α、水平位移R2和傾斜角θ[15],這4個變量如圖2所示.以3,9MeA:PHE二聚體為例說明4個參數,其中,水平位移R2的方向按照圖3虛線標明的角度進行移動.氨基酸殘基結構如圖3.選用可以形成二聚體穩定體系的4個變量值進行模型構建.結合文獻中4個變量的值,4種損傷腺嘌呤與氨基酸殘基的二聚體結構如圖4所示.
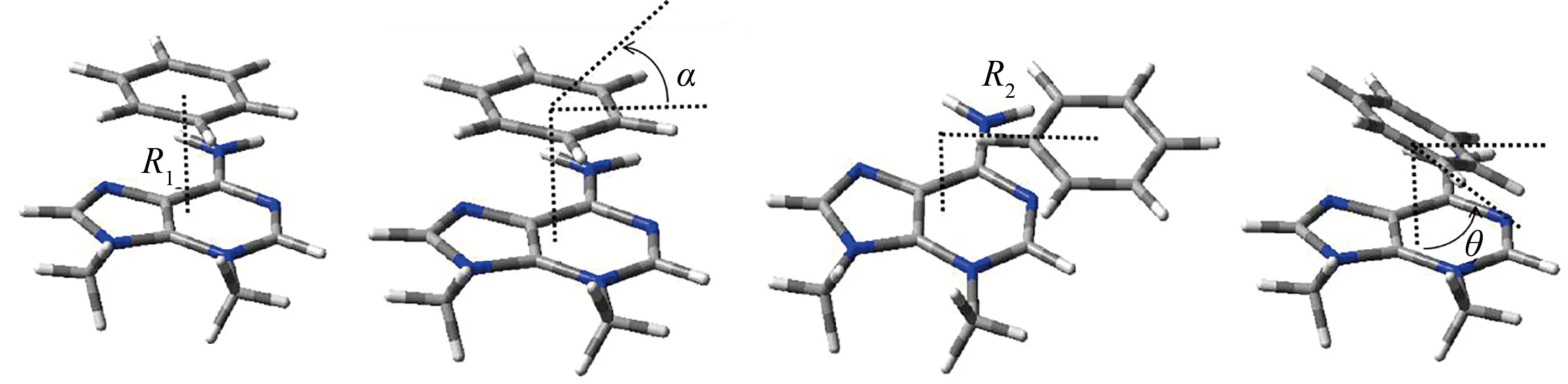
圖2 垂直位移R1、旋轉角α、水平位移R2和傾斜角θ變化示意圖

圖3 水平位移R2示意圖,虛線代表平移方向
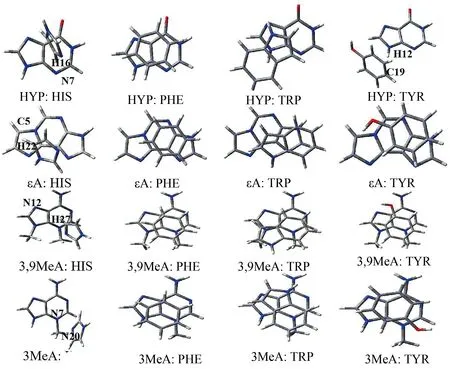
圖4 二聚體結構示意圖
2 計算細節與模型方法
2.1 計算細節
使用MP2/6-31G方法對受損腺嘌呤與氨基酸殘基組成的二聚體進行結構優化,使用MP2/aug-cc-pVDZ方法進行能量計算,計算的公式為
ΔE=En+Ea-Edi.
(1)
其中,Edi表示氨基酸殘基與受損腺嘌呤組成的二聚體的單點能,En表示受損腺嘌呤的單點能,Ea表示氨基酸殘基的單點能,ΔE代表氨基酸殘基與受損腺嘌呤之間的堆積能.
2.2 ABEEM極化力場模型
ABEEM極化力場除了設置原子位點,還定義了σ鍵、π鍵和孤對電子的位點,并且給出明確的電荷,這些位點的電荷隨著結構和環境的變化而變化,反映了體系的電荷轉移和由外部分子所引起的極化效應.
ABEEM極化力場與其他力場相比,最為突出的特色是靜電相互作用,采取庫侖相互作用形式,具體表達如式(2):
(2)
其中,q是原子位點、鍵位點及孤對電子位點的電荷,rij是任意兩位點i和j間的距離,參數kij是校正因子,它充分考慮了電子與原子核之間的屏蔽效應及鉆穿效應,具體表達為式(3).
(3)
其中,kHB是氫鍵擬合函數具體公式參見文獻[14],kπH/ππ是堆疊擬合函數,具體公式為式(4),k′,C,D,rπH/ππ是參數,RπH/ππ是H原子和π位點的距離,或者兩個分子中π位點之間的距離.
(4)
2.3 ABEEM極化力場參數調節
參數的調節是本工作的重點,也是最耗費時間的部分.模型分子包括4個損傷腺嘌呤、4個氨基酸殘基,16個損傷腺嘌呤和氨基酸殘基的堆疊二聚體.本工作主要調節相關π位點參數和堆疊擬合函數相關參數.π位點電荷參數通過線性回歸和最小二乘法優化,擬合HF/STO-3G[14]水平的電荷.堆疊擬合函數通過擬合MP2/aug-cc-pVDZ方法的堆疊能確定,相關參數列于表1.
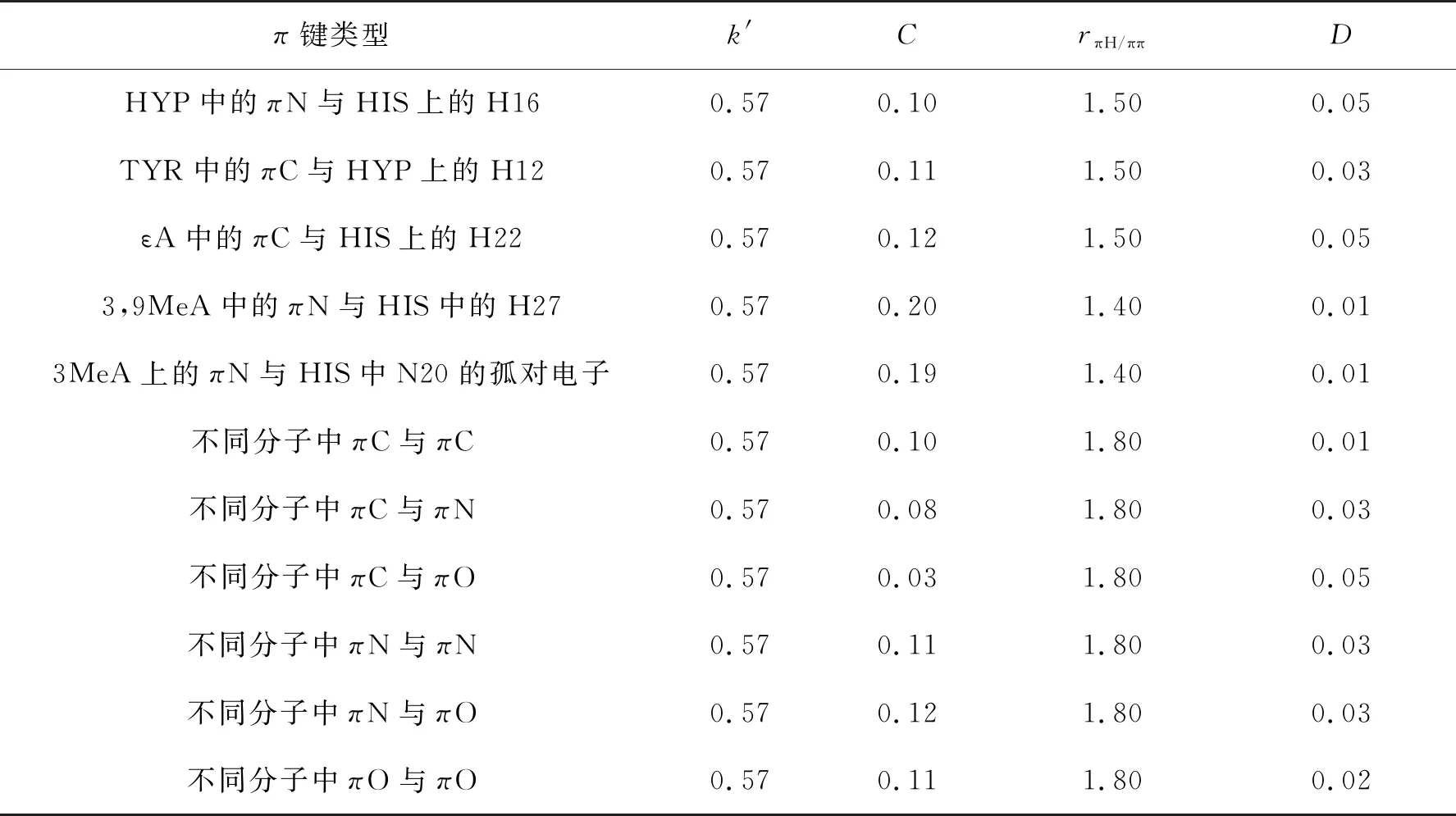
表1 堆疊擬合函數相關參數
3 結果與討論
3.1 幾何結構
用MP2/6-31G方法和ABEEM極化力場方法將幾何結構進行優化,得到的穩定結構的參數平均絕對偏差如下:HYP與氨基酸體系的R1、R2的偏差分別為0 , 0.002 5 nm,α、θ的偏差分別為0.25,0°;εA與氨基酸體系的R1、R2的偏差分別為0,0.01 nm,α、θ的偏差分別為2.25,0.5°;3,9MeA與氨基酸體系的R1、R2的偏差分別為0,0.025 nm,α、θ的偏差分別為1.25 , 0.5°;3MeA與氨基酸體系的R1、R2的偏差分別為0,0.005 nm,α、θ的偏差分別為0.5 , 0.5°.上述結果表明ABEEM極化力場可以很好地模擬生物體系的堆積作用.
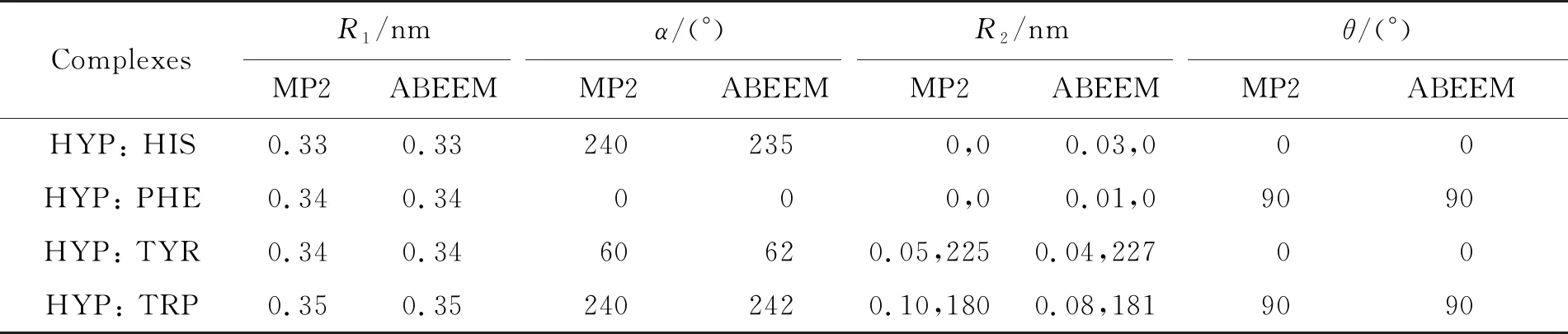
表2 HYP與氨基酸體系的4個最優參數
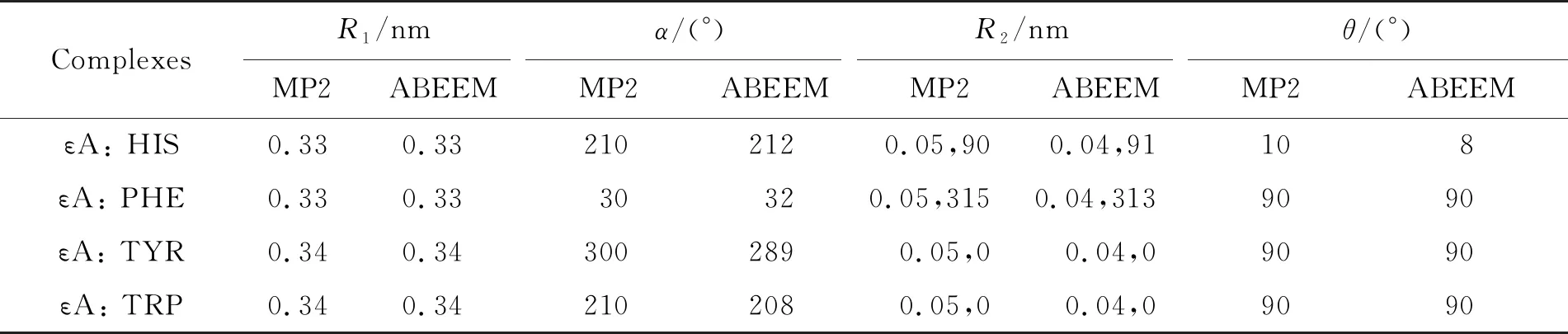
表3 εA與氨基酸體系的4個最優參數

表4 3,9MeA與氨基酸體系的4個最優參數

表5 3MeA與氨基酸體系的4個最優參數
3.2 電荷分布
比對HF/STO-3G方法下的電荷分布與ABEEM極化力場下的電荷分布發現,兩者的線性相關系數都在0.97以上,說明ABEEM極化力場可以快速、準確地計算大分子體系的電荷分布.
偶極矩是判斷電荷分布是否準確的物理量,將中性二聚體——HYP和εA分別與4種氨基酸形成的二聚體的偶極矩,在MP2/aug-cc-pVDZ和ABEEM極化力場下進行對比,二者的平均絕對偏差為0.39×10-30C·m,線性相關系數是0.98,說明ABEEM極化力場計算的電荷分布是合理的.只是斜率為0.747 3,說明ABEEM極化力場的偶極矩比MP2/aug-cc-pVDZ方法的偶極矩稍小.
3.3 堆疊能
用MP2/aug-cc-pVDZ和ABEEM極化力場計算出各損傷腺嘌呤、氨基酸單體與16種二聚體的單點能,可以發現:相同氨基酸下,各損傷堿基的堆疊能大小為3,9MeA>3MeA>εA>HYP,烷基化堿基的堆疊能要比中性受損堿基的堆疊能大,且隨著烷基的數量增多而增大,3,9MeA的堆疊能甚至是HYP的2倍.對比MP2/aug-cc-pVDZ方法和ABEEM極化力場的堆積能發現,能量平均絕對偏差為1.06 kJ/mol,二者的線性相關系數是0.99,非常接近于1,且斜率為1.019 8.可以看出,無論是偏差的數值還是線性相關系數均很好地說明了ABEEM極化力場能夠準確地模擬堆積相互作用.
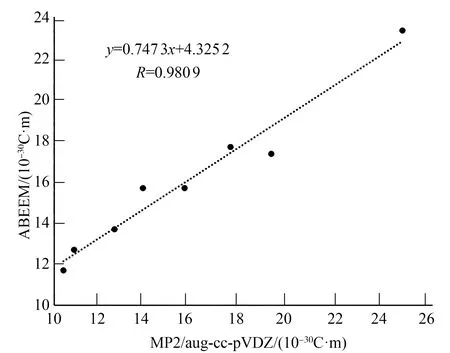
圖5 偶極矩線性相關圖
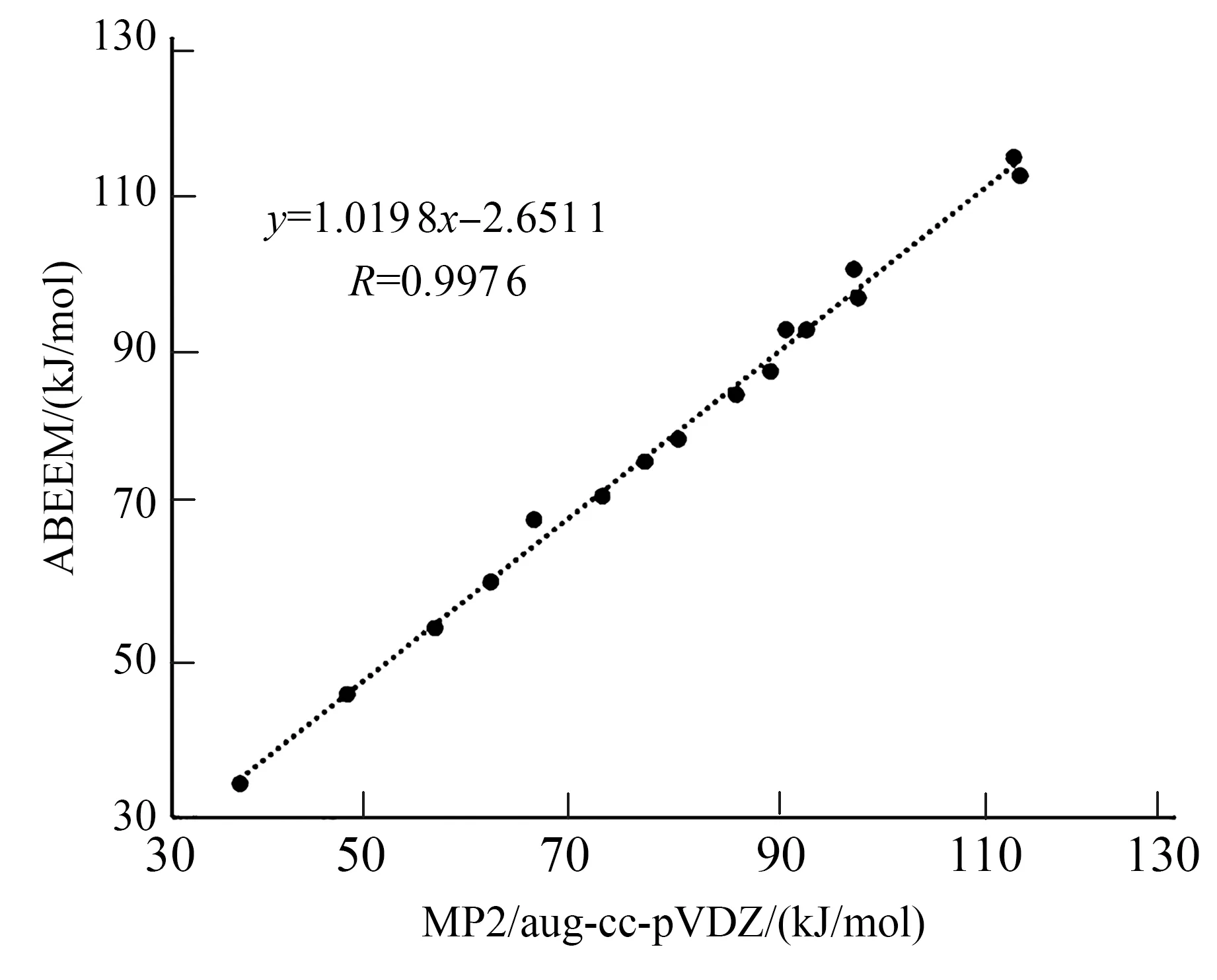
圖6 堆疊能線性相關圖
4 結 論
本文使用MP2/aug-cc-pVDZ//MP2/6-31G方法對HYP、εA、3,9MeA和3MeA分別與4種氨基酸殘基所形成的二聚體體系進行結構優化和能量的計算,將結果作為調節ABEEM極化力場π鍵位點參數的依據,本文發展的ABEEM極化力場的最突出貢獻就是添加了π鍵位點和堆疊作用函數,使模擬的堆積作用二聚體體系更加準確.將ABEEM極化力場計算得出的結果與量子力學結果進行對比,其電荷分布、偶極矩以及堆積能的線性相關系數分別是0.97、0.98和0.99,都非常趨近于1,有很好的擬合相關性,說明本工作發展的ABEEM極化力場可以精準地模擬氨基酸殘基與受損腺嘌呤間的堆積相互作用.并且用于這16種堆積二聚體體系的參數具有可轉移性,結合其考慮了分子間的靜電相互作用的優勢,ABEEM極化力場能夠很好地應用于DNA與蛋白質生物大分子體系為修復機理的研究提供理論基礎.
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
