離子液體和低共熔溶劑催化二氧化碳合成有機碳酸酯的研究進展
2022-04-12 03:54:30阮佳緯葉香珠陳立芳漆志文
化工進展 2022年3期
阮佳緯,葉香珠,陳立芳,漆志文
(華東理工大學化工學院,化學工程聯合國家重點實驗室,上海 200237)
全球氣候變暖已成為近年來的重大環境問題乃至政治問題。中國提出要在2030 年前實現碳達峰,2060年前實現碳中和,體現了中國作為負責任大國應對全球氣候變化的擔當,是挑戰,更是機遇。二氧化碳(CO)作為主要溫室氣體,雖是氣候變暖的要因,但也是豐富和可再生的碳一資源。基于此,國內外發展了具有應用前景的碳捕獲與封存(carbon capture and storage, CCS)和碳捕獲與利用(carbon capture and utilization,CCU)技術,以減緩氣候變暖,同時制備高附加值化學品。CO的捕集與資源化利用已經成為能源與環境領域的重要研究方向。
目前,以CO為原料可合成有機碳酸酯、甲醇、甲烷、尿素和淀粉等多種化學品。其中,有機碳酸酯應用廣泛,包括直鏈與環狀碳酸酯。碳酸二甲酯(DMC)是一種重要的直鏈碳酸酯,不僅是理想的汽油和柴油添加劑,也是低毒、低黏度的綠色溶劑。環狀碳酸酯在工業上可以用作極性非質子溶劑、鋰電池電解質組分和有機合成中間體。在合成路線方面,CO能與甲醇直接合成DMC,與環氧化物通過環加成合成環狀碳酸酯,這些符合“綠色化學”和“原子經濟”的化學反應被廣泛研究。
CO的熱力學穩定性和化學惰性限制了其轉化利用。為克服CO轉化的反應能壘,人們發展了多種應用于CO合成有機碳酸酯的催化體系,其中包括金屬鹵化物、金屬配合物、金屬有機骨架和氮化碳納米片等。但這些催化劑普遍存在反應條件苛刻、需要添加溶劑或助溶劑、成本高等缺點。離子液體(ILs)作為一種新型的綠色介質,具有結構可設計性、高催化活性和易分離特性等優勢,在催化CO合成有機碳酸酯方面備受關注。低共熔溶劑(DESs)被視為新一代ILs,是由一定化學計量比的氫鍵受體(HBA)和氫鍵供體(HBD)通過氫鍵作用形成的低熔點混合物。它不僅具有ILs 的優良性質,還具有合成簡單、價格低等優勢,近年來在CO轉化為有機碳酸酯反應體系中被深入研究。
本文綜述了近年來國內外對于ILs和DESs催化CO轉化為有機碳酸酯反應的研究進展,重點介紹了不同種類ILs和DESs的催化性能和作用機理。
1 CO2合成有機碳酸酯的反應機理
1.1 CO2與甲醇直接合成碳酸二甲酯
以CO和甲醇為原料直接合成DMC是CO高效轉化途徑之一。該反應受熱力學平衡限制,難以實現高轉化率。Kabra 等對該反應進行了熱力學分析,計算了常溫常壓下反應的標準生成焓Δ(-16.5kJ/mol)和標準吉布斯自由能Δ(+38.0kJ/mol),表明該反應是放熱反應,且在常溫常壓下不能自發進行;并通過實驗研究發現低溫高壓有利于提高DMC 收率。即使在高壓下,該反應平衡轉化率仍不高,需要設計高效催化體系和催化脫水耦合過程。而催化劑的酸性位和堿性位對CO和甲醇的活化起重要作用,兼具酸堿活性位的ILs 在該反應中的應用逐漸得到發展。
理清CO與甲醇的反應機理有利于開發高效催化體系,文獻主要結合實驗與量子化學計算開展研究。Casarin 等揭示了CO在反應中既可作為電子受體也可作為電子供體,認為CO的2π(LUMO)軌道可接受來自催化劑堿性位的電子,1π(HOMO)軌道則提供電子給酸性位。因此CO在催化劑表面的活化,一種可能是通過電子從催化劑轉移到CO,進而形成彎曲的CO來占據LUMO 軌道;另一種可能是電子從CO轉移到催化劑表面,CO轉變為CO,此時CO會保持線性構型。Tomishige 等證明了堿性位活化CO形成彎曲的陰離子,酸性位活化甲醇形成CH。Zhao 等提出了酸堿雙功能ILs 催化CO和甲醇直接合成DMC 的分子活化過程和反應機理(如圖1 所示):首先是堿性位作用,OH將電子轉移給CO,形成彎曲的CO;甲醇在堿性的OH作用下很容易生成CHO,隨后CHO親核進攻CO中帶正電荷的碳原子,形成碳酸單甲基取代陰離子CHOCOO;接著是氫鍵酸性位的作用,羥基和具有布朗斯特堿性的OH協同進攻甲醇,在甲醇和ILs 上的羥基間形成氫鍵網絡,導致甲醇O—H 鍵和C—O 鍵極化,形成CH;最后,活化的CHOCOO與CH+反應生成DMC。
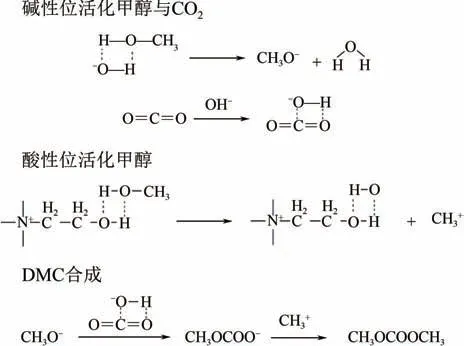
圖1 CO2與甲醇直接合成DMC的分子活化過程與反應機理[19]
1.2 CO2與環氧化物環加成反應合成環狀碳酸酯
目前工業上制備環狀碳酸酯的催化劑主要是季銨鹽和堿金屬鹵化物,由于催化活性較低,需要在高溫高壓下操作,每生產1噸環狀碳酸酯會排放約0.9噸CO,這反而造成CO凈排放。因此,開發高效催化劑尤為重要。近年來,ILs 在CO環加成反應中逐漸得到應用,多種新型ILs 在常溫常壓下可催化CO高效轉化。為指導催化劑的開發,文獻深入探討了反應機理。
目前用于該類反應的催化劑通常包含酸堿活性位,普遍接受的ILs 催化機理是:酸性官能團(路易斯酸如金屬離子等,以及HBD基團如—OH、—COOH 等)活化環氧底物,堿性官能團(鹵化物、羧酸鹽等)親核進攻環氧底物上位阻較小的碳原子,隨后CO被活化并插入親核進攻中間體,最后發生分子內重排,脫除親核基團,得到環狀碳酸酯,并完成催化劑再生。
Girard 等結合實驗和密度泛函理論(DFT)計算,提出了兩種可能的ILs 催化CO環加成反應路徑,同時探討了潛在的副反應(如圖2 所示)。在循環1中,路易斯酸中心(咪唑環2號位碳上的質子)通過與環氧化物形成氫鍵活化環氧底物(Ⅰ),由堿性中心鹵素原子親核進攻活化的底物,生成烷氧中間體(Ⅱ);隨后CO插入烷氧中間體形成非環狀碳酸鹽陰離子(Ⅲ),親核基團脫除,生成環狀碳酸酯并完成催化劑再生。需要注意的是,此路徑中可能發生副反應:異構化生成醛以及水合生成一元或二元醇。在循環2 中,由咪唑環-雜環卡賓結構與CO配位,形成加合物(Ⅳ);隨后該加合物親核進攻環氧底物(Ⅴ)生成烷氧基中間體(Ⅵ),最后通過分子內親核取代得到環狀碳酸酯。對循環1 進行理論計算,優化了反應物、中間產物、過渡態、最終產物的幾何構型,計算了過渡態最低能量,得到總反應放熱為17.35kcal/mol(1cal=4.1868J),證明了該催化反應路徑的熱力學可行性。
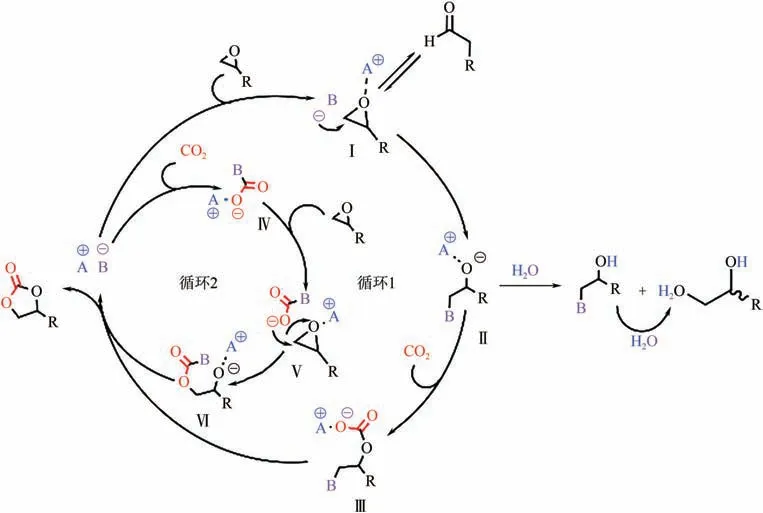
圖2 CO2與環氧化物環加成反應機理[24]
2 基于離子液體的CO2轉化
ILs通常指熔點低于100℃的有機鹽,由有機陽離子和有機或無機陰離子組成,具有不揮發、液程范圍寬、熱穩定性好、溶解能力強等常規有機溶劑無法比擬的優點。在CO轉化過程中,ILs 可以作為溶劑、CO吸收劑、CO活化劑、催化劑或助催化劑。文獻探究了ILs 的陽離子、陰離子、烷基鏈長、官能團以及溫度、壓力、催化劑用量等對催化活性的影響。
2.1 傳統離子液體
傳統ILs 是一類基礎ILs,包括咪唑類、吡啶類、季銨鹽與季膦鹽類等。國內外已經報道了大量用于催化CO轉化為有機碳酸酯的傳統ILs。
由于CO與甲醇直接合成DMC 反應受熱力學限制,及時脫除副產物水能推動化學平衡正向移動,因此當前研究主要對高效催化劑和脫水劑進行設計。Zhao 等合成了[CCIM][HCO],在室溫下將CO和甲醇合成DMC。他們發現該類ILs 會與CO形成加合物,且該加合物能結合副產物水生成咪唑碳酸氫鹽,因此該ILs 可同時用作催化劑和脫水劑(如圖3 所示),既突破化學平衡的限制,又簡化后續的脫水劑分離,實現CO和甲醇的高效轉化。Hu 等先通過咪唑環-雜環卡賓結構將CO進行化學活化,再將獲得的加合物與醇直接反應,在常溫常壓下制備了不同的直鏈碳酸酯,CO轉化率最高達40.2%,DMC 選擇性達99.9%;結合O同位素示蹤法和DFT 計算,揭示了在一鹵代烴和二鹵代烴存在下CO與甲醇轉化為DMC 的不同反應路徑,解釋了鹵代烴的烷基化效應以及推動化學平衡正向移動的作用。
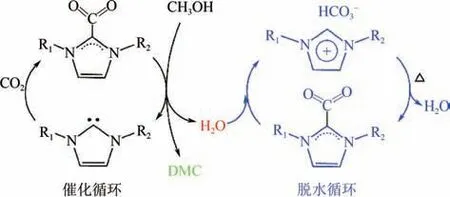
圖3 咪唑碳酸氫根ILs的催化與脫水耦合機理[31]
除了移除副產物水來突破化學平衡限制,Zhang 等在尿素兩步法醇解制DMC 反應中通入CO帶走氨氣并再生尿素以推動平衡正向移動。首先二醇基咪唑ILs 與尿素生成環狀碳酸酯中間體和氨氣,氨氣被通入的CO帶走;隨后通入甲醇與環狀碳酸酯進行酯交換反應,得到DMC并完成ILs的再生。在最優反應條件下,1mol尿素最多可以生成0.64mol DMC,其收率是尿素與甲醇直接反應的64倍。
傳統ILs 用于CO合成環狀碳酸酯也被大量報道。Anthofer 等合成了10種含不同取代基的咪唑溴鹽,其中1-五氟苯基-3-正辛基咪唑溴鹽在70℃、0.5MPa 下,催化環丙烷的轉化率最高達到91%,碳酸丙烯酯選擇性超過99%,且循環使用10次后,催化活性仍保持穩定。Xiao 等發現咪唑ILs 催化活性隨咪唑環酸性的增強而增加,這是因為強酸性有利于ILs 中氮相連的質子與環氧化物的氧原子之間形成氫鍵來活化底物;該ILs 循環使用5 次,在保持選擇性的同時,活性略有降低。Girard 等探究了一系列咪唑ILs 對CO的吸收能力,并將其用于環狀碳酸酯的合成。他們發現,適量水有利于提高環狀碳酸酯的收率,且無副產物二醇生成;但過量水會導致環氧底物開環和醇鹽水解,加速生成副產物二醇。
Toda等報道了一種由四芳基取代的季膦鹽和鹵素組成的、帶有布朗斯特酸性位和親核位的ILs(如圖4 所示);發現只有鄰位羥基取代的季膦鹽ILs 具有催化活性,且活性高,環狀碳酸酯收率能達到90%,而間位和對位取代的ILs沒有催化效果;還指出季膦鹽中芳基上的吸電子基團會降低其催化活性。Wu等報道了一系列四丁基膦的ILs,其中[BuP][2,4-OPym-5-Ac]在常溫常壓下反應20h,-亞烷基環狀碳酸酯收率達到91%,且對多種炔丙醇底物都具有良好催化活性。
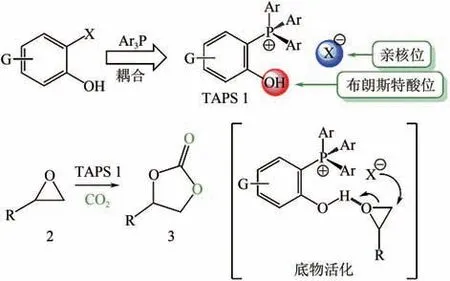
圖4 四芳基鹵素膦鹽作為環加成反應雙功能催化劑[37]
2.2 質子型離子液體
質子型ILs 是由等摩爾量的布朗斯特酸和布朗斯特堿構成的一種新型ILs。區別于其他ILs最主要的性質是其有質子從酸到堿的轉移,導致存在質子供體和質子受體,因此可構建強氫鍵網絡。此外,它還具有低成本、易制備和質子活性可調等優勢,使其在CO捕集和轉化方面被廣泛研究。
常見的有機強堿如二甲氨基吡啶(DMAP)、1,8-二氮雜二環[5.4.0]十一碳-7-烯(DBU)等常被用作質子型ILs 的質子受體,它們能與CO形成加合物從而活化底物。Roshan 等通過DFT 計算了有機堿催化CO與環氧底物環加成過程中形成的穩定中間體和過渡態,發現CO-堿-水之間形成的HCO能提高反應活性。Meng 等制備了一系列DBU 基質子型ILs(如圖5 所示),用于催化CO和環氧氯丙烷的環加成反應,其中f在常溫常壓下的模擬煙道氣(15%CO,85%N)中反應6h,環氧氯丙烷轉化率達92%;分析其傅里葉變換紅外光譜發現,與CO作用后的ILs在1975cm處出現了一個對應于氨基甲酸鹽的不對稱振動峰,意味著該ILs的烷氧基陰離子活化了CO。理論計算表明,使用催化劑f時反應活化能最低,最有利于開環;f與環氧底物過渡態中氫鍵長度最短,氫鍵作用最強,故活化底物能力最強。需要注意的是,該ILs循環使用4次后,環狀碳酸酯收率從99%降為77%,主要是環氧底物加成到ILs上導致活性位點的活性降低。
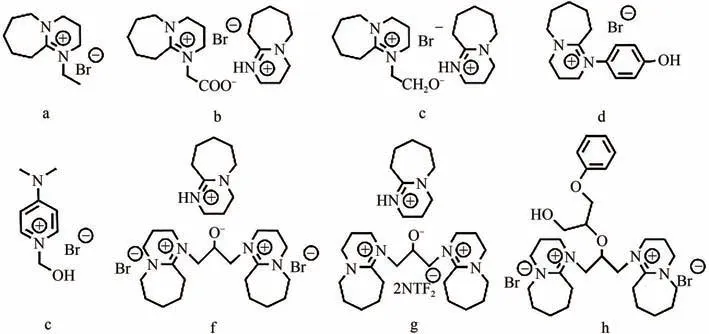
圖5 質子型ILs[42]
Mujmule 等研究了有機堿DBU、三乙胺、,-二異丙基乙胺等與咪唑基ILs 組成的二元催化體系,發現DBU 堿性強且活性氮附近空間位阻較小,易于活化CO。在120℃、2MPa下,[EVIMOH][Cl]/DBU 中反應1h,環氧丙烷轉化率為99.8%,產物選擇性超過99%。該體系中羥基官能團、咪唑環上2號位碳上的活性質子以及含N堿性位之間的協同作用能促進CO的吸收,增強反應活性;在循環使用5 次后催化活性無明顯降低。Zhang 等制備了DMAP基質子型ILs,其中[DMAPH][Br]在120℃、常壓下,針對模擬煙道氣反應14h,環氧苯乙烯轉化率為92%。他們認為質子型ILs 的有機堿陽離子上的正電荷離域增強了與環氧化物和CO的相互作用,從而提高催化活性。該ILs 對多種環氧底物都具有良好活性,且易回收。Chen 等則通過調控ILs 的堿度,使之能同時作為吸收劑和活化劑,在常壓下亞烷基碳酸酯收率達91%。
2.3 功能化離子液體
結構可調是ILs 最重要的特性。利用其結構可設計性,在陰陽離子上引入具有特殊功能的官能團可獲得功能化ILs。在CO轉化為有機碳酸酯的反應中,常見的功能化基團有羥基、羧基和氨基等。功能化ILs對CO表現出強吸收固定能力,在CO轉化方面也表現出比常規ILs更高的催化活性。
Wang等借助DFT計算,證明了在CO與環氧底物的環加成反應中,ILs 的羥基是關鍵活性位。Zhao等合成了含有不同羥基數量的功能化胺基和咪唑基ILs,在催化CO和甲醇直接合成DMC 反應中,芐甲基二羥乙基氫氧化銨具有最佳催化效果,證明了醇羥基和OH的協同作用對CO的固定和活化起關鍵作用,另外還發現陽離子上芐基取代基帶來的共軛效應有利于甲醇的羰基化反應。該ILs 在循環使用4次后,甲醇轉化率有明顯降低,可能是由于回收時的質量損失。Sun 等開發了羥基功能化ILs,在[HEMIM][Br]中,125℃、2MPa 下反應1h,環氧丙烷轉化率高達99.2%,碳酸丙烯酯選擇性達到99.8%。
Sun等還開發了羧基功能化的咪唑ILs用于催化CO與環丙烷的環加成反應,考察了氫鍵強弱和酸度對催化活性的影響,認為酸堿雙功能ILs 催化劑的強酸性能增強陽離子的開環能力,但也會削弱陰離子的親核性,最終降低碳酸丙烯酯產率。其循環使用5次后,催化活性無明顯下降,轉化率仍達97%,選擇性為99%。Xiao 等發現弱酸有利于提高催化活性,酸性過強時會形成強氫鍵而阻礙CO的插入,降低目標產物產率。Meng 等發現可以通過控制羧酸基ILs 的烷基鏈長來調節其在反應體系中的溶解度。在加熱條件下形成均相,從而表現出最大催化效率;反應結束后降溫,ILs 與產物分相,易于分離。
氨基功能化咪唑ILs 由于其活化和固定CO的能力,能有效催化CO與環氧化物的環加成反應。Chen等通過DFT計算指出,氨基功能化咪唑ILs 中咪唑環主要催化開環,質子化的胺基則穩定溴離子的親核進攻。Yue等在[APBIM][I]中,在120℃、1.5MPa 下反應1.5h,碳酸丙烯酯收率能達94.3%。他們指出,陽離子上較長的烷基鏈有助于提高ILs 的催化活性,而鹵素陰離子的催化活性順序為I>Br>Cl,這與Liu 等得到的結果一致。此外,氨基功能化ILs 既能與CO反應生成氨基甲酸鹽來活化CO,又能通過氨基上的質子與環氧底物的氧原子形成氫鍵來活化環氧底物,從而實現對CO和底物的雙重活化。Yue 等嘗試了不同種氨基酸作為陰離子的咪唑基雙功能ILs 在90℃、0.25MPa 下催化CO與環氧底物的環加成反應,反應12h后碳酸環氯丙烯酯最高收率達到99%。該氨基酸ILs在200℃下不會分解,循環使用4次后,催化活性略有降低。
傳統ILs 穩定性較好,但反應條件較苛刻,催化活性也偏低。質子型ILs 易制備、催化活性高,近年來已有在常溫常壓下仍具有高催化活性的質子型ILs 被報道,但其穩定性仍需提高。多種功能化ILs 能獲得超過95%的產物收率,但反應條件較苛刻,難以在溫和條件下催化轉化。此外,多數功能化離子液體黏度較大,工業化應用有所局限。
3 基于低共熔溶劑的CO2轉化
ILs因制備復雜、難生物降解、成本高等缺陷,不利于工業化應用。DESs是HBA與HBD在一定比例下通過氫鍵作用結合形成的低熔點混合物。常見的HBA 包括氯化膽堿、季銨鹽、季膦鹽等,HBD 包括醇、羧酸、酰胺、糖類等。DESs 具有廉價、易獲取、可生物降解、無毒等ILs不具備的優良性質,被認為是ILs 的潛在替代品,適合規模化生產。
DESs 的獨特理化性質使之在氣體吸收、液體分離、反應催化、電化學等領域表現出優異性能。在CO捕集方面,報道較多。Fu 等制備了DBU 與吡咯組成的DESs,發現實際吸收量遠大于理論吸收量,并證明DESs 解離產生的離子化程度決定其吸收能力。此外,有研究指出DESs 對CO的吸收能力與其酸度或堿度無關,而與其HBA 和HBD之間的相互作用強弱有關。
在DESs 用于CO化學轉化方面的研究尚處于起步階段。通過改變HBA 與HBD 的種類和配比,可以調控對特定反應物的氫鍵形成能力,提高DESs 的吸收能力與催化活性。目前,以氯化膽堿、季銨鹽與季膦鹽為HBA 構成DESs 催化CO與環氧底物反應的報道較多,有機堿和其他類型的HBA 較少。本文涉及DESs 的組成如圖6所示。
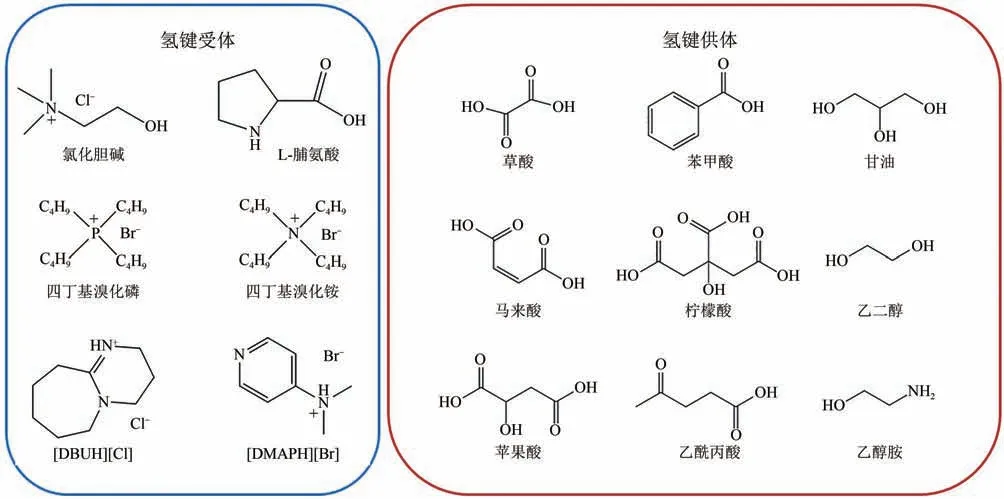
圖6 本文所涉及DESs的組成
3.1 氯化膽堿基低共熔溶劑
氯化膽堿無毒、生物相容性好、價格低廉,是組成DESs最常用的HBA。由其構成的DESs制備簡單,可生物降解,而且可以通過改變HBD 種類與配比來改變DESs的物理化學性質。
Tak等首次報道了在DESs中將螺環氧吲哚通過環加成合成新型螺環碳酸酯的策略,并以甘油、乙二醇、苯磺酸和尿素作為HBD 制備了氯化膽堿基DESs。在70℃、常壓下經過2h,在氯化膽堿/尿素組成的DESs催化下螺環碳酸酯產率能達到98%。Cheng等首次將氯化膽堿和ZnBr組成的路易斯酸性DESs用于CO環加成反應,在110℃、1.5MPa下反應1h,環狀碳酸酯收率高達99%,TOF 為494h。Liu 等開發了基于-羥基丁二酰亞胺的DESs,在碘化膽堿與-羥基丁二酰亞胺以1∶2摩爾比組成時,在室溫下反應10h,環氧丙烷轉化率達到96%。該DESs 可以通過萃取分離,且循環使用5 次催化活性無明顯降低。Dindarloo Inaloo 等制備了無水合金屬氯化物和氯化膽堿組成的DESs,將CO、胺和鹵代烴三組分偶聯成氨基甲酸酯,發現氯化膽堿和氯化鋅以1∶2的摩爾比組成的DESs具有最好的催化活性,室溫下反應2h 產物收率達95%。該DESs 循環使用6 次后催化活性略有降低,多種底物仍能獲得90%以上的氨基甲酸酯收率。
氯化膽堿還能與生物質衍生的有機酸和醇形成DESs。Vagnoni 等將乙二醇、甘油等醇類HBD 和草酸、檸檬酸、蘋果酸、馬來酸、酒石酸等羧酸類HBD與氯化膽堿、碘化膽堿等HBA制備的DESs用于CO與環氧底物環加成反應,發現碘化膽堿基DESs 在常壓下的催化活性普遍高于氯化膽堿基DESs,這歸因于I優秀的離去能力;HBA 和HBD在未形成DESs時的催化能力較形成DESs后有明顯下降,說明DESs 具有更完整的氫鍵網絡,活化底物能力更強。
3.2 季銨鹽和季膦鹽基低共熔溶劑
季銨鹽是工業上合成環狀碳酸酯常用的催化劑,由于催化活性低,需要對其進行優化。而形成DESs 是一種可行方式,既避免使用有機溶劑,又可以利用HBA與HBD的協同作用提高催化活性。
Wang 等開發了以脂肪族羧酸作為HBD,季銨鹽作為HBA 的DESs,在80℃、0.4MPa 下,環氧丙烷轉化率最高達98%,碳酸丙烯酯選擇性超過99%;發現pa值為3.7~4.9的脂肪族羧酸與四丁基溴化銨組成的DESs 催化活性最佳,底物轉化率均大于95%。Yingcharoen 等以不同pa 值的酚、一元和多元醇、羧酸、抗壞血酸等作為HBD,四丁基溴化銨、四丁基碘化銨等作為HBA,合成了DESs,考察了在CO與環氧氯丙烷的環加成反應中前20 min的初始反應速率常數,發現pa值為9~10.5的HBD 所形成的DESs 催化活性最高。Wang 等合成了不同羥基取代位置的吡啶和四丁基銨鹽組成的DESs,其中3-羥基吡啶與四丁基碘化銨形成的DESs 反應速率最快,環狀碳酸酯收率也最高,能達到95%。Liu 等將苯酚,苯胺,苯磺酸和鄰、間、對位氨基苯酚作為HBD,與四丁基溴化膦形成DESs,用于環氧底物環加成反應,在四丁基溴化膦與間氨基苯酚以1∶2摩爾比組成的DESs催化活性最高,其循環使用5 次,催化活性無明顯降低。Liu 等以四丁基溴化胺為HBA,與乙二醇、1,2-丙二醇、三甘醇、丙二酸、葵酸等HBD 制備了一系列DESs,用于環氧化大豆酸甲酯與CO的環加成反應,在最優化條件下反應10h,大豆酸甲酯收率達到95%。
季銨鹽與季膦鹽作為HBA,除了能與羧酸、醇類等形成DESs,也能與金屬鹵化物作用形成DESs。Sun等發現在ZnCl/PPhCHBr(摩爾比1/6)中,在120℃、1.5MPa下反應1h,環氧丙烷轉化率達到96%,TOF達4718.4h。Rehman等發現在季銨鹽與ZnBr組成的DESs體系中,鹵代季銨鹽中陰離子活性順序為Br>I>Cl>F。Br由于其優秀的親核能力和離去能力,表現出最高的催化活性。
3.3 有機堿基低共熔溶劑
有機堿作為HBA形成的DESs與有機堿基質子型ILs類似,組成DESs的HBD可以是布朗斯特酸,也可以是醇、醇胺等。此外還可以將有機堿基ILs與HBD以一定的摩爾比混合,得到DESs。
Sun 等研究了以有機堿DBU 和纖維素組成的DESs 催化CO與環氧底物的環加成反應,在120℃、2.0MPa 下反應2h 環氧丙烷轉化率為93%。對比實驗及原位紅外光譜,推斷DBU 和纖維素對CO與環氧底物的活化具有協同作用,并提出了可能的催化機理(如圖7 所示),纖維素活化底物,DBU 既進攻環氧底物,又與CO形成加合物活化CO,兩個中間體偶聯后DBU與纖維素脫除并完成再生,得到環狀碳酸酯。
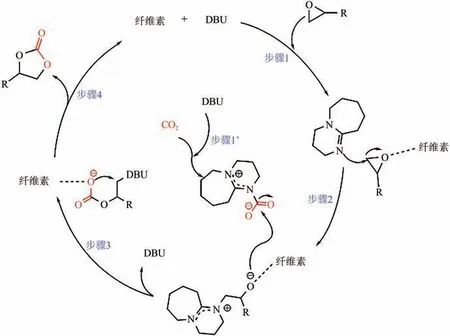
圖7 DBU-纖維素體系催化CO2環加成反應的作用機理[80]
Garcia-Arguelles 等以超強堿1,5,7-三疊氮雙環[4.4.0]癸-5-烯(TBD)和DBU為HBA,苯甲醇、乙二醇、甲基二乙醇胺等醇類為HBD,合成了DESs。發現提高HBD 比例可以提高對CO的吸收能力,而1∶1摩爾比的TBD與苯甲醇形成的DESs在100℃、1.2MPa 反應條件下,CO環加成反應中環狀碳酸酯收率最高達到98%。Yang等將質子型ILs 作為HBA,與尿素、乙醇胺、二乙醇胺(DEA)、甲基乙醇胺等HBD 形成DESs,在常溫常壓下,[DBUH][Br]-DEA(2∶1)中碳酸苯乙烯酯收率能達97%。
3.4 其他類型
除了上述組成,文獻還報道了氨基酸等其他類型的HBA 組成的DESs。Lyu 等以氨基酸為HBA,與二元酸合成了多種天然DESs,在130℃、1.2MPa下反應5h,碳酸丙烯酯收率高達98.6%。值得注意的是,這類DESs需要在共催化劑ZnBr的協助下才能實現高收率。Wang 等開發了由[BMIM][Cl]/硼酸/戊二酸組成的三元DESs 并將其用于催化CO與環氧丙烷的環加成反應,在配比為7∶1∶1時,碳酸丙烯酯收率最高能達98%。該三元DESs 催化體系循環使用5次,選擇性保持不變,但由于產物覆蓋了活性位點以及分離過程中DESs 的損失,底物轉化率有所降低。
DESs 在CO捕集與催化轉化方面已取得一定研究進展,但仍存在穩定性較差、后續分離困難等問題。DESs 作為二元或多元混合物體系,由于HBA 與HBD 的沸點差異,與有機溶劑和ILs 相比,在復雜體系中其分離更困難,用傳統的分離方法回收的DESs 可能因其損失難以恢復到原始組成。開發能獲得高純產品的催化劑和過程工藝,將更具有學術和應用價值。
4 結語
目前ILs 在CO與甲醇合成DMC、與環氧化物合成環狀碳酸酯反應中已獲得較高催化活性,但也存在反應條件苛刻、成本高、分離困難等缺點。高效ILs 催化劑通常具有以下結構:①兼具酸性位與堿性位,酸堿活性位能有效活化反應底物和CO,調控陰陽離子酸堿度能實現對不同反應體系的高效催化活性;②陰離子通常選取兼具強親核性和強離去能力的基團,如鹵素陰離子;③陽離子通常選取體積大、具有電子離域作用的基團以及大分子有機堿,如咪唑、吡啶、DBU、DMAP等常作為陽離子。
DESs被視為ILs的廉價替代品,對其研究雖起步較晚但發展迅猛。DESs 在催化DMC 合成中的研究還很少,在環狀碳酸酯合成中DESs 催化劑尚存在催化活性不高、反應底物種類較少等不足。在后續研究中,可以重點關注以下問題。
(1)DESs 在有機碳酸酯合成反應中的催化機理尚需深入研究,理清HBA 和HBD 與反應體系之間的分子相互作用有助于解釋DESs 結構與性能之間的關系。
(2)探索更多種類的HBA和HBD。根據ILs開發的經驗,開發基于多尺度計算或深度學習的計算機輔助設計方法,有助于針對特定反應體系篩選或設計出合適的DESs。
(3)開發能在溫和條件下,催化低濃度CO與甲醇或來自可再生資源的生物基環氧化物轉化為有機碳酸酯的DESs,更有利于環境與經濟可持續發展。
