基于GEO數據庫研究結腸癌的生物標志物
2022-04-28 08:41:10文雪梅夏俊凱張麗靜賈彥彥張文俊
中國現代醫生 2022年9期
文雪梅 夏俊凱 張麗靜 賈彥彥 張文俊

[摘要] 目的 通過生物信息學篩選出結腸癌中的關鍵基因,為結腸癌分子生物研究及早期診斷相關生物標志物篩選提供理論依據。方法 從GEO數據庫中選擇編號為GSE10950、GSE74602和GSE110224的基因芯片,利用R語言下載評估芯片質量,對樣本進行規范化處理并篩選出差異基因(DEG),通過TCGA下載結腸癌的表達數據,驗證DEG的準確性,通過DAVID在線網站對DEG進行GO分析和KEGG富集分析,通過STRING在線網站得到DEG的蛋白互作網絡,利用Cytoscape 軟件篩選出樞紐基因(hub基因),Oncomine數據庫從基因層面驗證hub基因的表達,cBioPortal數據庫分析hub基因在結腸癌中的突變情況,最后通過UALCAN及TCGA數據庫評估hub基因在結腸癌診斷方面的價值。結果 通過 GEO數據庫共篩選出結腸癌132個DEG,經TCGA數據庫驗證后最終得到131個DEG,其中表達下調DEG 78個,表達上調DEG 53個,通過STRING、Cytoscape篩選出10個hub基因,選取排名靠前的DTL等4個hub基因進一步分析,最終證明了DTL、CCNA2、BUB1、KIF23基因在結腸癌中的高表達并可能作為早期診斷結腸癌的標志物。結論 通過生物信息學分析研究篩選出結腸癌的關鍵基因,并證明蛋白編碼基因DTL、CCNA2、BUB1、KIF23有可能作為早期診斷結腸癌的生物標志物。
[關鍵詞] 結腸癌;生物信息學;GEO數據庫;TCGA數據庫;生物標志物
[中圖分類號] R735.3? ? ? ? ? [文獻標識碼] A? ? ? ? ? [文章編號] 1673-9701(2022)09-0001-07
Research on biomarkers of colon cancer based on GEO database
WEN Xuemei1? ?XIA Junkai1? ?ZHANG Lijing2? ?JIA Yanyan1? ?ZHANG Wenjun3
1.Xinhua Clinical College, Dalian University,Dalian? ?116021, China;2.Department of Gastroenterology, Xinhua Hospital Affiliated to Dalian University, Dalian? ?116021, China;3.Department of Anorectal Surgery, Xinhua Hospital Affiliated to Dalian University, Dalian? ?116021, China
[Abstract] Objective To screen out key genes in colon cancer through bioinformatics, and provide a theoretical basis for the molecular biology research of colon cancer and the screening of relevant markers for early diagnosis. Methods The gene chips numbered GSE10950, GSE74602, and GSE110224 were selected from GEO database. R language was used to download and evaluate the quality of the chip. The sample was standardized and the differential gene (DEG) was screened out. The expression data of colon cancer was downloaded through TCGA to verify the accuracy of DEG.GO analysis and KEGG enrichment analysis of DEG was performed through DAVID online website. The DEG′s protein interaction network was obtained through the STRING online website. The hub gene was screened out using the Cytoscape software. The expression of hub genes verified by Oncomine database. The mutation of hub gene in colon cancer was analyzed through cBioPortal database. And finally the value of hub gene in colon cancer diagnosis was evaluated through UALCAN and TCGA databases. Results A total of 132 DEGs for colon cancer were screened through the GEO database. After verification by the TCGA database, 131 DEGs were finally obtained. Ten hub genes were screened through STRING and Cytoscape. The top 4 hub genes, including DTL, were selected for further analysis. It was proved that DTL, CCNA2, BUB1, KIF23 genes were highly expressed in colon cancer and might be used as markers for early diagnosis of colon cancer. Conclusion This study screened out the key genes of colon cancer through bioinformatics analysis, and proved that the protein coding genes DTL, CCNA2, BUB1, KIF23 might be used as biomarkers for the early diagnosis of colon cancer.
[Key words] Colon cancer; Bioinformatics; GEO database; TCGA database; Biomarkers
結直腸癌是常見的消化道惡性腫瘤,其發病率、死亡率分別居全球惡性腫瘤的第4位和第2位[1],且仍處于上升階段[2]。結腸癌高死亡率的主要原因是缺乏早期癥狀,且臨床常用的腫瘤標志物缺乏診斷早期結直腸癌的能力,某些分子的表達水平又因人而異,在此情況下,早期篩查和診斷的難度明顯增加。因此,找到新的能有效診斷早期結直腸癌的標志物也就十分重要。
近年來,高通量測序技術及基因芯片研究受到醫藥領域的廣泛關注,一些數據庫含有大量樣本的特征為醫學研究的可靠性及可行性提供了一定的保證。通過對這些大數據的研究分析,可以從上萬個基因中篩選出在腫瘤發生發展中有重要作用的關鍵基因。本文通過生物信息學發現并研究一些關鍵基因在結腸癌中的表達情況,并進一步分析關鍵基因診斷早期結腸癌的潛力,現報道如下。
1 材料與方法
1.1 材料來源
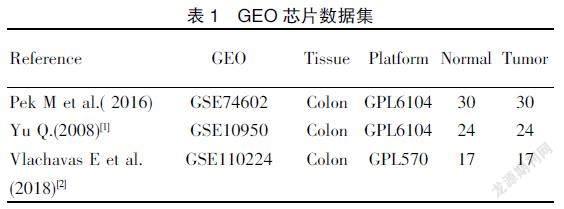
從公共基因芯片(gene expression omnibus,GEO)數據庫(http://www.ncbi.nlm.nih.g——ov/geo)選取表達譜芯片數據集GSE10950、GSE74602及GSE110224,芯片信息見表1。從癌癥和腫瘤基因圖譜(the cancer genome atlas,TCGA)數據庫(https://portal.gdc.cancer.gov/)選取結腸癌轉錄組數據,共包含41例正常樣本和480例腫瘤樣本。分析工具主要為R語言(R×64 4.02版本)及各類R包、Cytoscape(3.8.0版本)、Graphad Prism 7統計軟件、MedCalc? Application統計軟件(19.1.0版本)以及各類在線數據分析網站。
1.2 方法
1.2.1 篩選差異基因? 通過GEOquery R包[3]對選取的GEO芯片數據進行下載、預處理及規范化。對處理后的數據進行分析,設置篩選條件為校驗P值小于0.05(adj.P.value<0.05),表達變化倍數的絕對值大于2(|log2FC|>1),并利用limma R包[4]分別篩選出3個芯片數據集的表達差異基因(differentially expressed genes,DEG)。通過在線軟件draw Venn diagrams(http://bioinformatics.psb.uge--nt.be/webtools/Venn/)獲得3個數據集取交集后的重疊DEG。
1.2.2 驗證差異基因? 通過TCGAbiolinks? R包[5]下載TCGA數據庫中結腸癌的轉錄組數據,按照疾病部位為Colon,項目為 TCGA-COAD,數據種類為transcri ptome profiling,實驗種類為RNA-seq,工作流程的類型為HTSeq-counts進行數據下載。通過NCBI獲得人類基因組注釋文件GTF進行數據合并及注釋轉換。設置DEG篩選條件為:|log2FC|>1,adj.P<0.05,利用DESeq2 R包[6]分析整理并得到DEG,并通過draw Venn diagrams與GEO得到的重疊DEG合并,以驗證GEO數據庫得到的DEG。根據LogFC的大小使用pheatmap R包[7]繪制最終所得DEG的聚類熱圖。
1.2.3 差異表達基因的功能富集分析? 用于注釋可視化和集成發現的數據庫(database for annotation, visualization and integrated discovery,DAVID)(版本6.8)是一種在線數據庫( https: //David.ncifcrf.gov/),該數據庫集成了大量生物數據和相關分析工具,提供了系統而全面的生物信息高通量基因表達的功能注釋信息[8]。 將最終所得DEG導入DAVID數據庫中,進行DEG的基因本體論(gene ontology,GO) 注釋、京都基因和基因組百科全書(kyoto encyclopedia of genes and genomes,KEGG)途徑分析,研究DEG在細胞組分(cellular component,CC)、分子功能(molecular func-tion,MF)、生物學過程(biological process,BP)及參與信號途徑方面的富集情況。設定 P<0.05 為差異有統計學意義。
1.2.4 篩選樞紐基因相互作用基因(或蛋白質)檢索工具STRING數據庫(search tool for the retrieval of interacting genes,STRING)是一種在線工具(http://string-db.org/),旨在評估和整合蛋白質-蛋白質相互作用(PPI)信息,如物理和功能關聯,迄今為止,STRING 11.0版涵蓋了來自2,031個生物體的9,643,763個蛋白質[9]。通過STRING數據庫獲得DEG蛋白質互作(protein-protein interaction ,PPI)網絡,再將 PPI 網絡導入 Cyto-scape 軟件進行可視化分析,并利用插件 cytoHubba 篩選出樞紐基因(hub) 基因。
1.2.5 驗證hub基因? Oncomine數據庫(http://www.oncomine.org)是一種癌癥微陣列數據庫和基于Web的數據挖掘平臺,旨在激發全基因組表達分析的新發現,并比較大多數主要類型癌癥與各自正常組織中的轉錄組數據[10]。通過Oncomine數據庫分析hub基因在常見腫瘤中的表達情況。并選擇相應數據集,下載hub基因在結腸癌與正常結腸組織中的表達數據,利用Graphad Prism 統計軟件分析并繪制表達箱線圖,驗證hub基因在結腸癌中的表達情況。
1.2.6 分析hub基因的基因突變率? cBioPortal數據庫(https://www.cbioportal.org/)主要用于探索多維度癌癥基因組數據,以可視化分析基因、樣本和數據庫類型。使研究者可以交互探索不同樣本、基因、通路在遺傳學上的改變,并與臨床數據相結合[11]。通過cBioPortal數據庫選取合適的結腸癌數據集,分析hub基因在結腸癌中的突變率,研究在結腸癌中hub基因的表達變化是否因基因本身發生突變所致,以此推測hub基因在結腸癌中的表達穩定性。
1.2.7 評估hub 基因對結腸癌的診斷價值? UALCAN數據庫(http://ualcan.path.uab.edu/index.html)是一個交互式Web資源,使研究人員能夠快速輕松地分析來自TCGA數據庫的數據[12]。利用UALCAN數據庫獲得hub基因在結腸癌不同分期的表達情況,確認hub基因是否在早期結腸癌中出現表達升高的情況,然后整理TCGA獲得的基因表達數據,并利用統計軟件MedCalc? Application(19.1.0版本)得到hub基因的受試者工作特征曲線(receiver operating characteristic curve,ROC)及曲線下面積(area under curve,AUC),評估hub用于診斷結腸癌的潛力。
1.3 統計學分析
本研究差異基因的統計學分析通過R軟件進行,并采用非參數檢驗中的Mann-Whitney檢驗進行統計分析。hub基因驗證的統計學分析通過Graphad Prism 統計軟件,并采用非配對t檢驗進行分析。ROC曲線繪制及AUC計算通過MedCalc Application進行。所有分析均采用雙尾檢驗,α=0.05 為檢驗水準,P<0.05 為差異有統計學意義。
2 結果
2.1 DEG的篩選及驗證
通過R語言對選取的3個GEO芯片數據集的各表達譜進行分析,對數據集的各樣本表達譜進行規范化處理,按篩選條件通過limma R包篩選出各芯片數據集的DEG,并取交集后,共得到132個重疊DEG(封三圖1A),其中表達上調DEG 53個,表達下調DEG 79個。通過對自TCGA下載的結腸癌數據進行分析,共得到上調DEG 2863個,下調DEG 2552個。與GEO所得重疊DEG取交集后,排除GEO數據庫篩選得到的下調基因腺苷A3受體(adenosine A3 receptor,ADORA3),其余131個DEG均符合差異表達基因標準(封三圖1B),并通過pheatmap R包繪制最終所得DEG在各個數據集中的聚類熱圖(封三圖1C)。
2.2 DEG的GO分析和KEGG 途徑分析
通過DAVID 進行DEG的GO 分析(封三圖2)和 KEGG 途徑富集分析(表2)。GO分析結果顯示,DEG主要富集在細胞外泌體(extracellular exosome)、細胞表面(cell surface)及頂端質膜(apical plasma membrane)等細胞外部區域(封三圖2CC);主要參與細胞增殖(cell proliferation)、細胞外基質的組織(extracellular matrix organization)、氯離子跨膜轉運(chloride transmembrane transport)以及基因表達的正向調控(positive regulation of gene expression)等生物學過程(封三圖2 BP);主要參與轉運活性(transporter activity)、蛋白激酶結合(protein kinase binding)、生長因子活性(growth factor activity)、結構分子活性(structural molecule activity)、鈣離子結合(calcium ion binding)等分子功能(封三圖2 MF)。KEGG 途徑分析顯示,補體及凝血級聯(complement and coagulation cascades)、氮代謝(nitrogen meta-bolism)、白細胞跨內膜遷移(leukocyte transendothelial migration)、細胞周期(cell cycle)、NF-κB信號通路(NF-kappa B signaling pathway)等是DEG的主要富集信號途徑(表2)。
2.3 差異表達基因蛋白質互建網絡分析
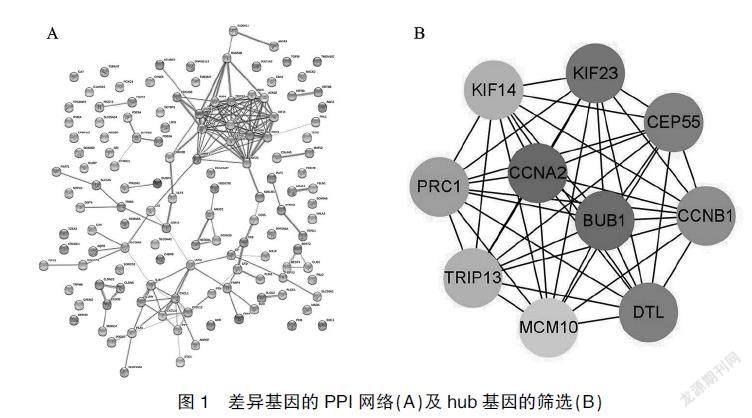
將最終得到的131個重疊DEG輸入STRING在線網站后得到PPI網絡(圖1A),將所得PPI信息下載后導入Cytoscape軟件中,利用 cytoHubba 插件篩選 hub基因并進行可視化,分別按MCC、Degree等12種計算方法進行分析,得到驅動蛋白家族成員23(kinesin family member 23,KIF23)等10個hub基因,為了便于后續深入分析,選取KIF23、有絲分裂檢查點絲氨酸/蘇氨酸激酶(BUB1 mitotic checkpoint serine/threonine kinase,BUB1)、細胞周期蛋白A2(cyclin A2,CCNA2)、無齒E3泛素蛋白連接酶同源基因(denticleless E 3 ubiquitin protein ligase homolog,DTL) 4個排名靠前的蛋白編碼基因進一步分析(圖3B)。
2.4 驗證hub基因在結腸癌中的表達
基于Oncomine數據庫分析4個hub基因在多腫瘤中的表達趨勢,結果顯示4個hub基因在腫瘤中多呈高表達狀態(圖2A)。進一步選擇Ki Colon[5]、Sabates-Bellver Colon[6]兩個結腸癌數據集進行分析,結果顯示相對于正常結腸組織,4個hub基因在結腸癌中明顯高表達(圖3)。
2.5分析hub基因的基因突變率
通過cBioPortal數據庫選取Kishore Guda等[13]、 Suhas Vasaikar等[14]的2項結腸癌數據集(CaseCCC、 PNAS 2015、CPTAC-2 Prospective、Cell 2019),共包括來自139例結腸癌患者的139份樣本。分析發現,BUB1等4種基因在結腸癌中的基因突變率均較低,最高的為BUB1,但也僅為4.0%(圖4),說明DTL等4種基因在結直腸癌中通過自身突變致癌的可能性低,其高表達可能是受到上游基因調控的結果,間接證明DTL等基因在結腸癌中的表達具有較好的穩定性。
2.6 hub 基因對結腸癌的診斷價值評估
通過UALCAN數據庫得到來自TCGA數據庫的315例臨床資料,分析 hub基因在結腸癌不同分期中的表達情況,結果顯示4個hub基因在結腸癌早期即出現了明顯的表達升高情況,并有隨著疾病進展表達逐漸降低的趨勢(圖 5)。通過MedCalc得到4個基因的ROC及AUC,結果顯示4個hub基因的AUC均大于0.9(圖2B),說明DTL等4種基因具有潛力可能作為結腸癌的診斷標志物。
3 討論
本文從GEO數據庫選取了3個結腸癌的芯片數據集,通過取交集的形式初步篩選出了132個在結腸癌中的差異表達基因,并進一步通過TCGA的結腸癌數據驗證了所得DEG的準確性,最終得到131個DEG,其中上調的DEG 53個,下調的DEG 79個。GO分析顯示這些DEG主要參與相關分子功能和生物學過程,KEGG分析提示DEG主要在細胞周期、NF-κB信號通路等信號途徑富集,而這些生物功能或通路均與腫瘤的發生發展有密切聯系。為了進一步找到hub基因,將得到的131個DEG導入到STRING在線網站并得到PPI網絡,將該PPI網絡導入到Cytoscape中,通過cytoHubba 插件的12種計算方法篩選后,得到hub基因。本文選取其中排名靠前的DTL等4個hub基因進一步分析。通過Oncomine數據庫發現,DTL等4個hub基因在多種腫瘤中明顯高表達且較穩定,亦驗證了其在結腸癌中的高表達。為了研究4個hub基因診斷早期結腸癌的潛力,首先通過UALCAN在線數據庫分析來自TCGA數據庫的315例樣本發現,DTL、KIF23、BUB1、CCNA2基因均在結腸癌的早期出現明顯高表達。此外,通過cBioPortal數據庫證實DTL等4種基因在結腸癌中的基因突變率偏低,說明DTL等4種基因表達具有較高的穩定性,符合診斷生物標志物的特點。最后,通過計算ROC曲線及AUC發現,這4個hub基因的AUC均大于0.9,證明它們的診斷早期結腸癌的準確率較高。
DTL又稱CDT2、DCAF2 或 RAMP,其在細胞周期、DNA復制及基因組穩定過程中發揮重要作用[16]。有研究發現,DTL在多種腫瘤中高表達,并被視為一種促癌基因[17]。Chen等[18]在實驗中發現,抑制DTL的表達能進一步抑制微管相關蛋白TPX2的表達,并認為DTL是通過癌細胞衰老誘導治療肝癌的潛在新靶基因。但在結腸癌中,尚無研究說明DTL的具體作用機制。CCNA2是一種高度保守的細胞周期蛋白,在控制細胞周期中起關鍵作用,CCNA2常在腫瘤中發揮促癌作用[19]。據Hung等[20]研究報道,在乳腺癌中,CCNA2缺失會導致蛋白激酶ERK活性受損和乳腺腫瘤細胞轉移減弱,在肝細胞癌中,CCNA2的表達通過激活PI3K/AKT促進腫瘤細胞的遷移、侵襲和轉移[21],CCNA2還能通過整聯蛋白αVβ3信號傳導途徑促進非小細胞肺癌的侵襲和遷移[22]。Gan等[23]在實驗中發現,在結腸癌中,CCNA2在癌組織中的表達高于正常組織,并且CCNA2敲低可以通過削弱細胞周期進程并誘導細胞凋亡顯著抑制結腸癌細胞的生長。
BUB1被認為是一種癌基因。據Piao等[24]發現,BUB1在胰腺癌組織中顯著過表達,且表達與胰腺癌的腫瘤大小相關。在膠質母細胞瘤中,通過抑制BUB1的表達出現癌細胞增殖減弱及腫瘤生長延遲現象[25]。生物信息學研究顯示,BUB1在乳腺癌及肝癌等腫瘤中高表達并具有可能作為靶向治療的標志物[26-27],在乳頭狀腎細胞癌中也得到同樣的結論[28]。有研究表明,BUB1的種系突變會增加結直腸癌的患病風險,但這種突變在結腸癌中并不常見,且這種說法還未得到充分證明[29-30]。KIF23已被證明可在體外驅動微管運動,并能與絲氨酸/蘇氨酸激酶家族(Aurora家族)的蛋白激酶相互作用,在有絲分裂步驟中起關鍵作用[31]。在乳腺癌中,KIF23水平升高與癌相關核調節蛋白ANCCA在腫瘤中的水平相關,且與ER陽性乳腺癌患者的無復發生存率低相關[32]。還有研究顯示,KIF23在肺癌中表達升高,是潛在的分子標記[33],同BUB1一樣,有研究報道KIF23在腫瘤中的水平升高可能是由于染色體的額外復制導致[34]。在結腸癌中,KIF23可能受到長鏈非編碼RNA UCA1的調控而發揮促癌作用[35]。目前針對DTL等基因表達升高的機制尚存在爭議,有研究者認為這是由于自身基因突變導致[29-30,34]。
總之,本文將生物信息學方法、高通量測序技術及基因芯片和大數據分析結合起來,發現并驗證了DTL、KIF23、BUB1、CCNA24個基因不僅在結腸癌早期出現中高表達且較穩定,同時基因突變率偏低,符合診斷生物標志物的特點并證明在早期結腸癌診斷中的準確率較高,因此有可能作為早期診斷結腸癌的生物標志物。且這4個基因在結腸癌中的高水平表達更多的是受到上游基因的調控導致,要進一步研究DTL等基因在結腸癌中的作用及作用機制,需重點關注其上游的調控基因。
[參考文獻]
[1]? ?Jiang X,Tan J,Li J,et al. Dact3 is an epigenetic regulator of wnt/beta-catenin signaling in colorectal cancer and is a therapeutic target of histone modifications [J].Cancer Cell, 2008,13(6):529-541.
[2]? ?Vlachavas EI, Pilalis E, Papadodima O, et al. Radio genomic analysis of f-18-fluorodeoxyglucose positron emission tomography and gene expression data elucidates the epidemiological complexity of colorectal cancer lands-cape[J].Comput Struct Biotechnol J,2019,17(177-185):178-186.
[3]? ?Davis S,Meltzer PS.Geoquery:A bridge between the gene expression omnibus(geo) and bioconductor[J].Bioinfor-matics, 2007,23(14):1846-1847.
[4]? ?Ritchie ME,Phipson B,Wu D,et al. Limma powers differ-ential expression analyses for rna-sequencing and micr-oarray studies[J].Nucleic Acids Research,2015,43(7):e47.
[5]? ?Colaprico A, Silva TC, Olsen C, et al. Tcgabiolinks: An r/bioconductor package for integrative analysis of tcga data[J].Nucleic Acids Research, 2016,44(8):e71.
[6]? ?Love MI,Huber W,Anders S.Moderated estimation of fold change and dispersion for rna-seq data with deseq2[J]. Genome Biol, 2014,15(12):550-553.
[7]? ?Kolde R. Pheatmap: Pretty heatmaps. R package version 1.0.12. 2019.
[8]? ?Huang DW,Sherman BT,Tan Q,et al. The david gene functional classification tool: A novel biological module-centric algorithm to functionally analyze large gene lists [J]. Genome Biol, 2007,8(9):R183.
[9]? ?Szklarczyk D, Morris JH, Cook H, et al. The string database in 2017: Quality-controlled protein-protein association networks, made broadly accessible[J].Nucleic Acids Research, 2017,45(D1):D362-D368.
[10]? Rhodes DR, Kalyana-Sundaram S, Mahavisno V, et al. Oncomine 3.0: Genes, pathways, and networks in a collection of 18 000 cancer gene expression profiles[J]. Neoplasia, 2007,9(2):166-180.
[11]? Cerami E,Gao J,Dogrusoz U,et al. The cbio cancer genomics portal: An open platform for exploring multidi-mensional cancer genomics data[J].Cancer Discov, 2012, 2(5):401-404.
[12]? Chandrashekar DS,Bashel B,Balasubramanya SaH, et al. Ualcan: A portal for facilitating tumor subgroup gene expression and survival analyses[J].Neoplasia,2017,19(8):649-658.
[13]? Guda K,Veigl ML,Varadan V,et al. Novel recurrently mutated genes in African American colon cancers[J].Proc Natl Acad Sci USA,2015,112(4):1149-1154.
[14]? Vasaikar S,Huang C,Wang X,et al. Proteogenomic analysis of human colon cancer reveals new therapeutic opportunities[J].Cell,2019,177(4):1035-1049.
[15]? Cheung WM, Chu AH, Chu PW, et al. Cloning and ex- pression of a novel nuclear matrix-associated protein that is regulated during the retinoic acid-induced neuronal differentiation[J].The Journal of Biological Chemistry, 2001, 276(20):17083-17091.
[16]? Slenn TJ, Morris B, Havens CG, et al. Thymine DNA glycosylase is a crl4cdt2 substrate[J].The Journal of Biolo-gical Chemistry, 2014,289(33):23 043-23 055.
[17]? Kobayashi H,Komatsu S,Ichikawa D,et al. Over expr-ession of denticleless e3 ubiquitin protein ligase homolog (dtl) is related to poor outcome in gastric carcinoma[J]. Oncotarget, 2015,6(34):36 615-36 624.
[18]? Chen YC,Chen IS,Huang GJ, et al. Targeting dtl induces cell cycle arrest and senescence and suppresses cell growth and colony formation through tpx2 inhibition in human hepatocellular carcinoma cells[J].Onco Targets Ther, 2018,11:1601-1616.
[19]? Yang F,Gong J,Wang G, et al. Waltonitone inhibits pro-liferation of hepatoma cells and tumorigenesis via fxr-mir-22-ccna2 signaling pathway[J].Oncotarget,2016,7(46):75 165-75 175.
[20]? Hung YH, Huang HL,Chen WC,et al. Argininosuccinate lyase interacts with cyclin a2 in cytoplasm and modulates growth of liver tumor cells[J].Oncology Reports,2017,37(2):969-978.
[21]? Gopinathan L, Tan SLW, Padmakumar VC, et al. Loss of cdk2 and cyclin a2 impairs cell proliferation and tumor-igenesis[J].Cancer Research, 2014,74(14):3870-3879.
[22]? Ruan JS,Zhou H,Yang L,et al. Ccna2 facilitates epithe-lial-to-mesenchymal transition via the integrin αvβ3 signaling in nsclc[J].Int J Clin Exp Pathol,2017,10(8):8324-8333.
[23]? Gan Y,Li Y,Li T,et al. Ccna2 acts as a novel biomarker in regulating the growth and apoptosis of colorectal cancer[J].Cancer Manag Res, 2018,10:5113-5124.
[24]? Piao J,Zhu L,Sun J,et al. High expression of cdk1 and bub1 predicts poor prognosis of pancreatic ductal adeno-carcinoma[J].Gene, 2019,701:26-30.
[25]? Yu H,Zhang S,Ibrahim AN,et al. Serine/threonine kinase bub1 promotes proliferation and radio-resistance in glioblastoma[J].Pathol Res Pract,2019,215(8):152 508.
[26]? Yang K,Gao J,Luo M.Identification of key pathways and hub genes in basal-like breast cancer using bioinfor-matics analysis[J].Onco Targets Ther,2019,12:1319-1331.
[27]? Yang WX,Pan YY,You CG. Cdk1,ccnb1, cdc20, bub1, mad2l1,mcm3,bub1b, mcm2,and rfc4 may be potential therapeutic targets for hepatocellular carcinoma using integrated bioinformatic analysis[J].Biomed Res Int, 2019, 2019:1245 072.
[28]? Gao Z,Zhang D,Duan Y,et al. A five-gene signature predicts overall survival of patients with papillary renal cell carcinoma[J]. PLoS One,2019,14(3):e0211 491.
猜你喜歡
中國醫藥導報(2017年1期)2017-03-15 17:37:13
南水北調與水利科技(2017年1期)2017-02-27 23:39:11
中國中藥雜志(2016年21期)2017-02-16 14:10:12
中國中藥雜志(2016年21期)2017-02-16 13:04:42
西南國防醫藥(2016年7期)2016-12-01 06:01:15
中國科技博覽(2016年8期)2016-04-25 06:02:18
中國科技博覽(2016年8期)2016-04-25 05:58:03
中國衛生標準管理(2015年1期)2016-01-14 03:41:26
河南醫學研究(2014年3期)2014-02-27 14:51:48
沈陽醫學院學報(2014年1期)2014-02-16 06:19:24
