持久性隆起性紅斑伴再生障礙性貧血一例并文獻復習
2022-05-17 03:45:48陳文杰孫勇虎劉永霞張福仁
中國麻風皮膚病雜志 2022年7期
陳文杰 孫勇虎 劉永霞 劉 紅 張福仁
山東第一醫科大學附屬皮膚病醫院(山東省皮膚病醫院),山東省皮膚病性病防治研究所,山東濟南,250022
1 資料與方法
1.1 臨床資料 患者,男,12歲。因“發現血象異常5年余,軀干及四肢皮疹1個月余”就診,曾于外院診斷為再生障礙性貧血,予環孢素治療后有效,好轉出院。皮膚科查體:雙肘伸側可見形狀不規則膚色隆起性斑塊(圖1a),臀部及雙膝伸側、雙大腿屈側見散在大小不等圓形或類圓形膚色斑塊,邊緣較中央隆起,無紅腫破潰,質中,皮溫不高,無觸痛(圖1b)。
皮膚組織病理(圖2):(右肘部)表皮角化過度,棘層輕度增厚,真皮內部分血管壁纖維蛋白樣變性,血管周圍較多中性粒細胞、少許淋巴細胞浸潤。實驗室檢查:血常規:血紅蛋白75 g/L, 紅細胞計數2.23×1012/L,白細胞計數3.09×109/L,中性粒細胞計數1.08×109/L,血小板計數86×109/L,CRP 2.02 mg/L。貧血相關檢測:可溶性轉鐵蛋白受體0.67 mg/L,鐵蛋白408.6 ng/mL,結合珠蛋白2.10 g/L,轉鐵蛋白1.62 g/L。骨髓穿刺(骨髓細胞形態學檢查報告):M:骨髓增生活躍,粒∶紅為0.58∶1。粒系增生減低,形態大致正常。紅系增生極度活躍,中晚幼紅細胞比例均明顯增高,形態大致正常。淋巴系增生明顯減低,其中幼稚淋巴細胞約占0.5%,淋巴細胞比例明顯減低,發現30個巨核細胞,血小板散在。免疫分型:紅細胞CD55、 CD59表達未見異常,白細胞CD55、CD59表達強度輕度下降。Coombs’試驗、生化、抗核抗體譜、凝血系列等均正常。
診斷:1、持久性隆起性紅斑;2、再生障礙性貧血。
治療:口服甲潑尼龍片12 mg,每日1次,治療后1個月,皮損變平,部分消退,可見色素沉著(圖3)。
1.2 文獻回顧 以“持久性隆起性紅斑”與“erythema elevatum diutinum”為關鍵詞,在知網和Pubmed搜索2000年至2022年1月持久性隆起性紅斑的病例報道,篩選了國內33篇,國外96篇文獻。
2 結果
2.1 基本信息 共137例患者,男72例,占52.6%,女65例,占47.4%;年齡最小5歲,最大85歲,平均50.71歲。
2.2 皮損臨床特征 該病好發部位為肢端與關節伸側,主要發病部位為手部82例,占59.9%;肘部63例,占46.0%;足部52例,占38.0%;膝部44例,占32.1%;下肢泛發35例,占25.5%;臀部27例,占19.7%;上肢泛發17例,占12.4%;另有少量病例于頭、頸、軀干等部位發病。對合并或不合并系統性疾病的EED患者,發現兩者之間在發病部位、皮疹的泛發程度、自覺癥狀等方面無明顯差異。
2.3 合并疾病 137例患者中單純表現為EED的患者83例,占60.6%,合并有其他疾病的54例,占39.4%,其中感染性疾病18例,占13.1%;血液系統疾病18例,占13.1%;自身免疫性疾病17例,占12.4%;血液系統腫瘤7例,占5.1%;其他系統腫瘤3例,占2.2%。見表1。
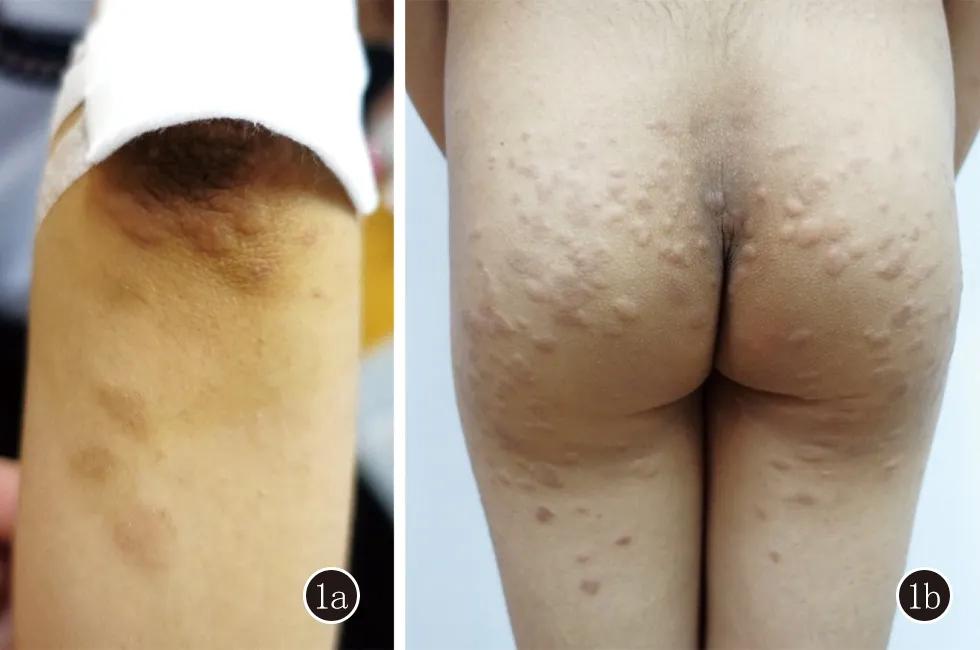
圖1 1a:肘部可見形狀不規則膚色隆起性斑塊;1b:臀部見散在大小不等圓形或類圓形膚色斑塊,邊緣較中央隆起
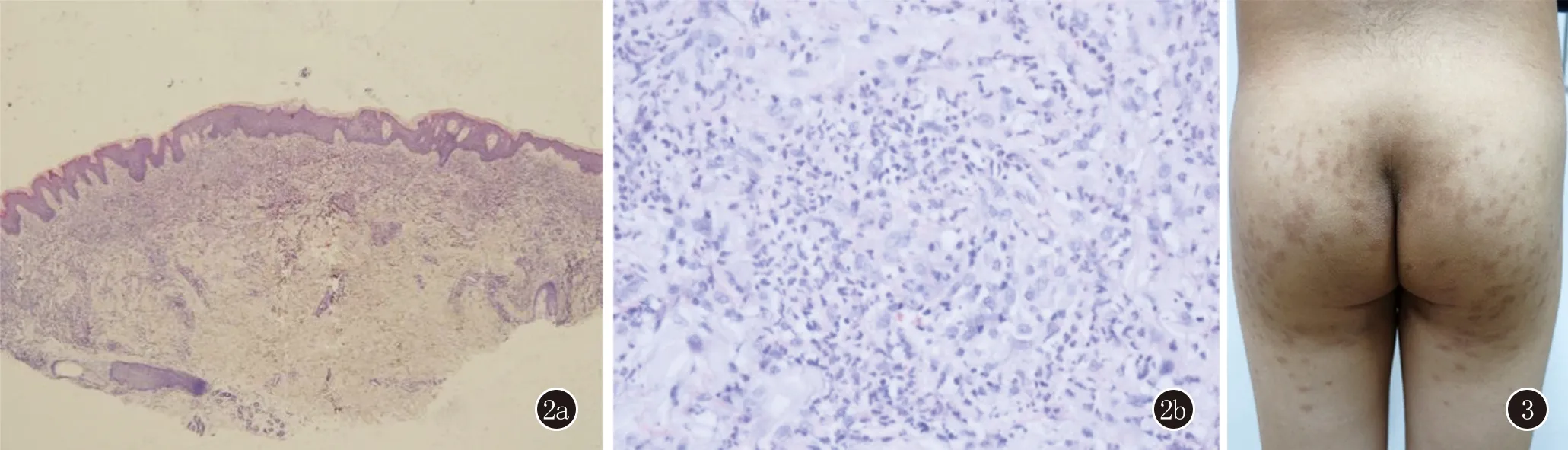
圖2 表皮角化過度,棘層輕度增厚,真皮內部分血管壁纖維蛋白樣變性,血管周圍較多中性粒細胞、少許淋巴細胞浸潤(HE,2a:×40;2b:×400) 圖3 治療1個月后,皮損變平,部分消退,可見色素沉著
3 討論
持久性隆起性紅斑(EED)是一種罕見的皮膚白細胞破碎性血管炎,常表現為紫紅色或紅棕色的丘疹、斑塊或結節,由Hutchinson[1]于1888年首次報道。EED的病因尚不明確,但被認為與感染、血液病或自身免疫性疾病的循環免疫復合物在血管壁沉積有關,從而導致炎癥級聯反應,使血管壁受損[2]。因此,EED除皮膚表現外,常常報道合并多種疾病,既往報道包括HIV感染、肝炎病毒感染、鏈球菌感染、克羅恩病、類風濕性關節炎、血液系統疾病和腫瘤等。
為了探討EED和系統性疾病的關聯,我們進行了系統的文獻回顧,發現39.4%的EED患者合并其他系統性疾病,其中副蛋白血癥的合并率最高。對合并或不合并系統性疾病的EED臨床特征進行了統計學分析,發現兩者之間在發病部位、皮疹的泛發程度、自覺癥狀等方面無明顯差異。
我們重點關注了EED合并血液系統腫瘤的情況,在診斷為非霍奇金淋巴瘤、急性髓系白血病、B細胞淋巴瘤的患者中[3-5],EED的皮損都先于腫瘤出現,并且在早期大多被檢測出有單克隆丙種球蛋白。非霍奇金淋巴瘤、B細胞淋巴瘤與另一例肺淋巴上皮瘤樣癌的患者,其EED的皮損與腫瘤的治療相平行,皮損隨著相對應的腫瘤治療(放療、化療等)而消退。EED對全身抗腫瘤治療有反應,而對局部治療沒有反應,也表明EED是副腫瘤綜合征的一種表現,對潛在疾病的成功治療可能利于EED癥狀的消除[6]。另外,有12例(8.8%)EED患者合并有副蛋白血癥,其中IgA副蛋白血癥有9例。該指標提示有可能發展為多發性骨髓瘤[7],值得臨床醫生關注。而在多發性骨髓瘤的患者中,EED皮膚征象的出現往往預示著可能轉化為白血病、 “原始細胞危象”等相關的不良預后[4]。因此,臨床上需要及時監測這類患者的血液學變化,免疫固定電泳也可以用于副蛋白血癥的初步診斷。

表1 137例患者合并疾病情況
本文報道的EED患者,合并有再生障礙性貧血,在國內外系首例合并報道。目前對于EED的發病機制,多認為是免疫復合物沉積在真皮血管壁,從而導致白細胞破碎性血管炎。既往曾有文獻報道10例EED患者中均出現了中性粒細胞IgA ANCA陽性,提示EED的皮膚血管炎可能與IgA ANCA激活中性粒細胞有關[8,9],即使血清IgA正常,也會出現IgA ANCA陽性的結果[10]。因此,可以認為IgA ANCA與EED密切相關。在再生障礙性貧血患者中,也有報道一例患者合并有ANCA相關性血管炎[11],免疫復合物在血管壁的沉積會導致發病。因此,我們推測再生障礙性貧血與后續EED發生存在共同的機制,但由于數據有限,尚需要有更多的證據支持。鑒于EED與血液系統疾病的強關聯性,以及EED在血液疾病的基礎上出現往往預示著不良的預后,并且常規治療常無效或容易反復,對于此類患者應該告知其相關風險,及時關注系統疾病受累情況,警惕其進一步進展為血液系統腫瘤。
