基于鋅配位的石墨烯/偶氮苯光熱材料
2022-05-25 06:07:16高文超馮奕鈺
功能高分子學報 2022年2期
高文超, 馮奕鈺, 封 偉
(天津大學材料科學與工程學院,天津 300072)
21世紀以來,日益增加的能源需求和突出的環境問題,使探索開發和使用可再生能源的需求變得越來越迫切[1,2]。太陽能作為清潔能源,其開發利用技術備受關注。具有光致可逆異構化特性的光響應性分子是實現高效可控光熱化學存儲與利用技術的關鍵,其中偶氮苯衍生物以其合成簡便、成本低、順反異構循環穩定性好等特點受到青睞[3-5]。
偶氮苯的發現可以追溯到19世紀中期,當時只被用作染料工業中的合成著色劑[6]。20世紀30年代,人們發現偶氮苯具有光異構化性質[7]。偶氮苯有兩種構型:反式構型和順式構型,它們之間存在一定的能級差。在紫外光照射下,偶氮苯可由低能量狀態的反式構型轉變為高能量狀態的順式構型,并將光能轉化為化學能并儲存在氮氮雙鍵(-N=N-)中。在一定的外場刺激下,偶氮苯會回復到低能態的反式構型并釋放熱量。偶氮苯在傳感器[8]、致動器[9]、非線性光學儀器、光學數據存儲[10]和能源/生物材料[11]等不同領域的研究取得了極大的進展。但是,偶氮苯能量密度低的缺點極大地限制了其進一步發展。
改善偶氮苯性能的方法主要有2種:(1) 設計并調整偶氮苯的結構[12,13];(2) 與其他分子(包括不同最大吸收峰波長的偶氮苯[14]、其他種類光熱分子[15]、聚合物[5,16]、相變材料[17,18]、碳材料[19~21]和其他材料[22])共混或鍵接。研究表明,與納米模板復合是設計兼具高可回收性、熱穩定性能源密集型太陽能熱燃料的有效策略。Grossman團隊[19]通過將偶氮化合物與單壁碳納米管(SCNTs)結合制備偶氮/碳納米管雜化材料,提高了偶氮的能量密度和半衰期。本課題組[20]通過偶合反應將偶氮苯接枝到還原氧化石墨烯(rGO)上,制得了一種能量密度為138 W·h/kg(約496.8 J/g)的雜化材料。除此之外,可以考慮將具有較高鍵能和良好動態可逆性的金屬配位鍵[23,24]引入偶氮苯基光熱材料,配合異構化過程實現兩種能量轉換協同作用的儲熱方式。
本文通過將4-硝基-4′-氨基偶氮苯(Azo)接枝到rGO上制得AGO,然后引入ZnCl2與AGO產生陰陽離子相互作用,得到石墨烯/偶氮苯(AGO/Zn)復合材料。石墨烯模板上偶氮苯的光異構化機制和離子相互作用機制,是通過復合熱釋放機制實現能量密度提高的新嘗試。通過傅里葉變換紅外吸收光譜等表征方式,證明AGO/Zn制備成功。差示掃描量熱分析結果表明,該方法大幅提高了AGO/Zn復合材料的能量密度(約為AGO能量密度的1.69倍)。
1 實驗部分
1.1 原料和試劑
Azo、石墨(595 μm):分析純,北京伊諾凱有限公司;氯化鋅(ZnCl2):分析純,天津弘鋒偉力科技有限公司;N,N-二甲基甲酰胺(DMF):超干溶劑,天津凱瑪特化工科技有限公司。
1.2 測試與表征
傅里葉變換紅外光譜(FT-IR)儀:德國Bruker光譜儀器公司TENSOR II型;X射線光電子能譜(XPS)儀:英國Kratos公司Axis Supra型,使用Mg Kα(1 250 eV)的激發源,測試功率450 W,真空度保持在10?8~10?9Torr(1 Torr=133 Pa),樣品質量 5 mg;場發射掃描電子顯微鏡(FESEM):日本日立公司 S8020 型,在液氮冷卻下進行測試,加速電壓5.0 kV,樣品5.0 mg;差示掃描量熱(DSC)儀:美國TA公司TAQ20型,在N2下進行測試,測試溫度范圍0~300 ℃,升溫速率10 ℃/min。
1.3 實驗步驟
1.3.1 氧化石墨烯(GO)的制備與還原 通過Hummers法制備氧化石墨烯[25,26];在油浴80.0 ℃條件下還原處理10 h制得rGO,標定質量濃度至8.25 mg/mL。
1.3.2 AGO的制備 AGO的合成示意圖見圖1。量取60.0 mL標定好的rGO-H2O分散體系加入到250 mL圓底燒瓶A內的100.0 mL乙醇中,在0~5 ℃冰浴反應并攪拌物料。稱取276.0 mg NaNO2溶于5.0 mL蒸餾水中,逐滴滴入到A中(約滴加5 min),反應1 h。稱取606.0 mg Azo溶于500 mL圓底燒瓶B內的30.0 mL乙醇中,同樣將B轉移到0~5 ℃冰浴中并攪拌物料。將A中的產物逐滴滴入到B中(約滴加20 min),在0~5 ℃反應4 h。去離子水洗1遍,乙醇洗3遍。上述過程重復2遍。在最后一次乙醇抽干前,分3次加入50.0 mLDMF,抽至剩20.0 mL液體。將產物轉移到500 mL容量瓶中,配制成質量濃度為0.25 mg/mL的溶液備用。
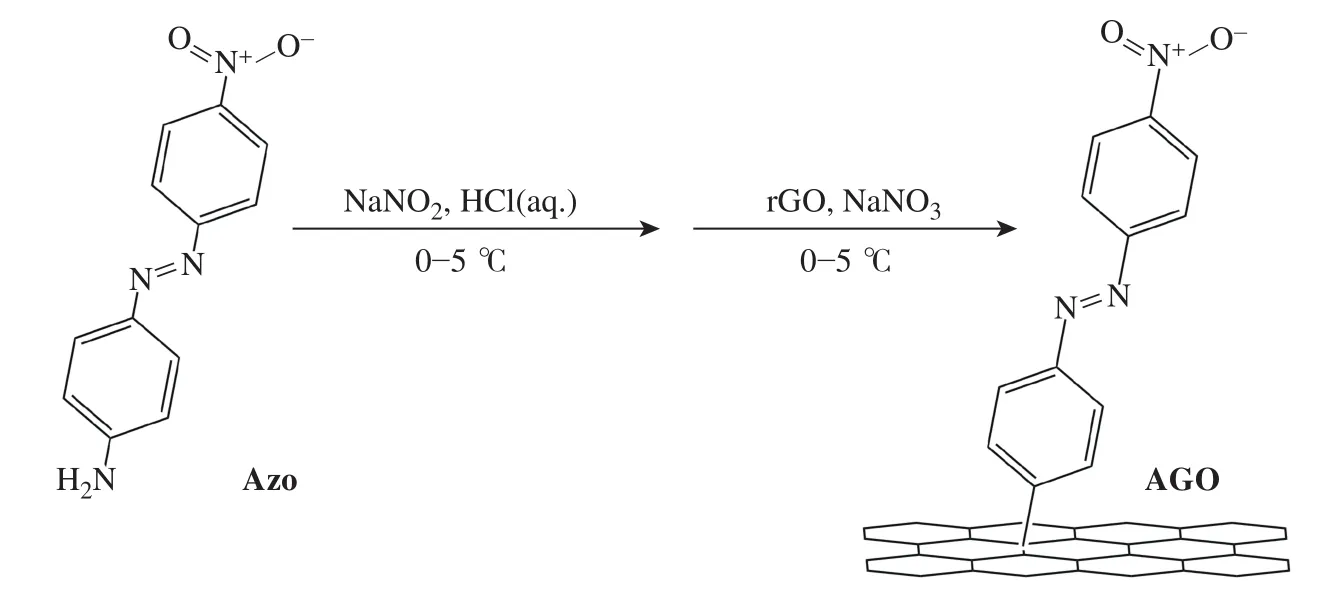
圖1 AGO 的合成Fig.1 Synthesis of AGO
1.3.3 AGO/Zn的制備 稱取27.3 mg的ZnCl2溶于5 mL樣品瓶內的2.0 mL超干DMF中,經超聲處理10 min,使其完全溶解。量取10.0 mL、0.25 mg/mL的AGO的DMF分散液于2個15 mL樣品瓶中,分別加入ZnCl2的DMF溶液和2.0 mL超干DMF,搖勻靜置,觀察其反應現象。
2 結果與討論
2.1 制備現象描述與分析
圖2為AGO/Zn的制備(即離子相互作用的形成)與空白組的對比圖,空白組的設置是為了排除其他因素的干擾。ZnCl2與AGO均勻混合,靜置20 min后,溶液中產生肉眼可見的微小顆粒;靜置60 min后,微小顆粒相互聚集形成明顯的絮狀物,且均勻分散在溶液中;靜置180 min后,絮狀物間相互聚集導致其空隙減小至肉眼觀察不到,開始沉淀;靜置240 min后,沉淀物下沉至樣品瓶中部,樣品瓶中呈現出兩個體積相似但顏色對比鮮明的兩部分;靜置600 min后,沉淀降至底部,體積不再變化。整個過程中,空白對照組并無明顯現象。

圖2 AGO/Zn的制備示意圖Fig.2 Schematic diagram of preparation of AGO/Zn
上述過程中,前期實驗組與對照組均無明顯現象的原因可能是,ZnCl2和AGO較緩慢地形成相互作用。在0~60 min時間段,與空白對照組相比,剛加入ZnCl2后實驗組無明顯現象的原因是相互作用形成較慢,僅局部片層之間通過離子相互作用連接,片層間距相對較大,未形成片層的堆積,在宏觀上不能為肉眼察覺;在20~60 min時間段,由于大量AGO/Zn形成導致片層有效連接和分子熱運動導致片層碰撞堆積,AGO/Zn聚集成能被肉眼觀察查到的絮狀物;在180~600 min時間段,在離子相互作用、分子熱運動和重力作用影響下,形成沉淀物。沉淀的形成說明AGO/Zn的重力大于浮力,AGO/Zn的密度大于溶液密度。與20 min的懸浮狀態相比,AGO/Zn的密度從與溶液密度等值的狀態轉變為大于溶液密度的狀態。由分析可知,AGO/Zn密度變化是由片層連接和堆積導致的。
2.2 FT-IR分析
圖3(a)為AGO/Zn和AGO的FT-IR對比圖。AGO和AGO/Zn分別在1 572 cm?1和1 581 cm?1處出現了硝基(-NO2)的反對稱伸縮振動峰,在1 339 cm?1和1 343 cm?1處出現了-NO2的對稱伸縮振動峰,在1 642 cm?1和1 640 cm?1處出現了氮氮雙鍵(-N=N-)的特征峰,印證了Azo成功接枝到rGO上。與沒有離子相互作用的AGO相比,AGO/Zn在429 cm?1處出現了Zn-O的特征峰[27],從而進一步佐證了金屬離子與AGO相互作用的形成。
2.3 XPS分析
圖3(b)是AGO/Zn和AGO的XPS譜圖。AGO和AGO/Zn在XPS譜圖中于282、397、529 eV處出現了C1s、N1s、O1s的特征峰,而AGO/Zn的XPS圖譜中在1 021.7 eV處出現了Zn2p的特征峰[27],且經過分峰得到AGO/Zn在1 022 eV處Zn-O的特征峰,從而證明Zn2+與AGO之間形成了相互作用。
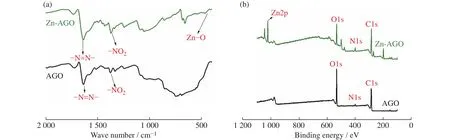
圖3 AGO/Zn的(a) FT-IR 光譜和(b)XPS能譜Fig.3 (a) FT-IR spectra and (b) XPS pattern of AGO/Zn
2.4 微觀形貌分析
圖4是rGO(圖4(a))和AGO/Zn(圖4(b, c))的FESEM照片。rGO片層較薄,呈微褶皺狀,片層間距較大[28];而AGO/Zn片層變厚,表面彎曲,片層間產生堆積。同時,對比圖4(b)和圖4(c)可知,在視野增大的情況下片層堆積尤為明顯,進一步佐證了前面的觀點。

圖4 樣品的FESEM照片Fig.4 FESEM images of samples
2.5 熱性能分析
圖5(a)是AGO 和AGO/Zn 在紫外光(365 nm)照射1 h后的DSC圖譜。與AGO的能量密度(299.1 J/g)相比,AGO/Zn的能量密度(370.4 J/g)大大提高,Zn2+的引入明顯提高了AGO的能量密度。紫外光(365 nm)照射時間對AGO/Zn 能量密度的影響如圖5(b)所示。隨著照射時間延長,能量密度呈現增長趨勢,但隨著照射時間的持續增加,增長幅度減小,這也與文獻[28]報道相一致。值得一提的是,在紫外光(365 nm)照射4 h后,AGO/Zn的能量密度達到最大值504.2 J/g,約為AGO能量密度的1.69倍。
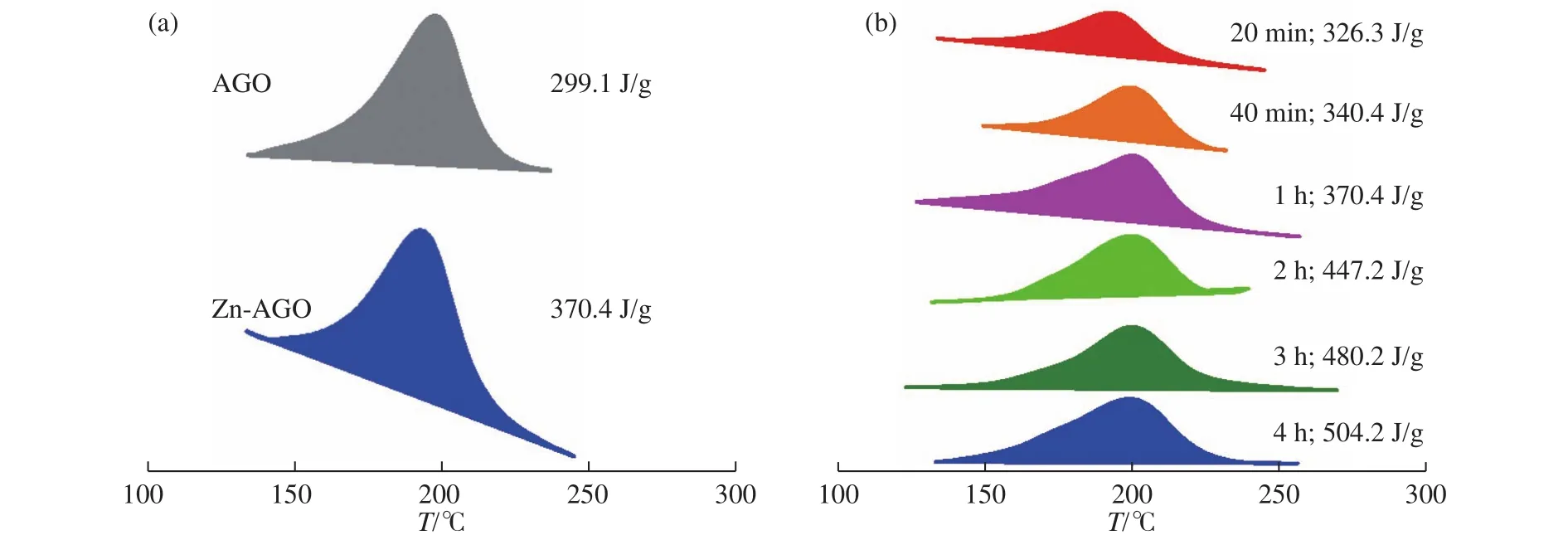
圖5 (a) AGO 和 AGO/Zn,(b) 不同紫外(365 nm)照射時間的 AGO/Zn 的 DSC 曲線Fig.5 DSC curves of (a) AGO and AGO/Zn, (b) AGO/Zn under different irradiation time of UV (365 nm)
3 結 論
(1)將Azo接枝到rGO上,并通過反應使之與ZnCl2產生相互作用成功制得AGO/Zn。
(2)在紫外光(365 nm)照射4 h后,AGO/Zn的能量密度達到最大,約為AGO能量密度的1.69倍。
(3)通過協同兩種儲熱機理,基于鋅離子作用制得石墨烯/偶氮苯光熱材料的能量密度顯著提高。
