UPLC-MS/MS法測定食用植物油中乙基麥芽酚含量的不確定度評定
2022-06-07 21:00:06李宣張瀠元黃永橋
食品安全導(dǎo)刊 2022年4期
李宣 張瀠元 黃永橋






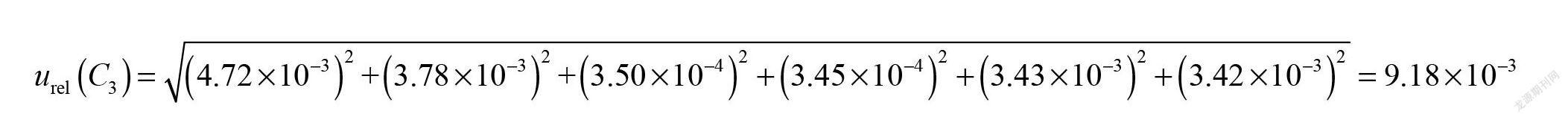


摘 要:依據(jù)BJS 201708規(guī)定的檢測方法,建立數(shù)學(xué)模型,評定超高效液相色譜-串聯(lián)質(zhì)譜法測定食用植物油中乙基麥芽酚含量的不確定度。通過對檢測過程中各環(huán)節(jié)不確定度因素的來源進(jìn)行分析并對其進(jìn)行評定,計(jì)算合成不確定度。結(jié)果表明,測定過程中不確定度的來源主要是標(biāo)準(zhǔn)溶液配制和標(biāo)準(zhǔn)曲線擬合;研究測得植物油中乙基麥芽酚的含量為95.6 μg/kg,其擴(kuò)展不確定度為4.3 μg/kg(k=2,95%置信區(qū)間)。研究可反映測量結(jié)果的置信度和準(zhǔn)確性,為日常實(shí)際檢測工作提供技術(shù)參考。
關(guān)鍵詞:超高效液相色譜-串聯(lián)質(zhì)譜法;食用植物油;乙基麥芽酚;不確定度
Uncertainty Evaluation of Determination of Ethyl Maltol in Edible Vegetable Oil by UPLC-MS/MS
LI Xuan1, ZHANG Yingyuan2*,HUANG Yongqiao1
(1.Guizhou Testing Technology Research and Application Center, Guiyang 550014, China; 2.Guizhou Academy of Testing and Analysis, Guiyang 550014, China)
Abstract: According to the detection method specified in BJS 201708, a mathematical model was established to evaluate of the uncertainty of the determination of ethyl maltol in edible vegetable oil by ultra-high performance liquid chromatography tandem mass spectrometry. The sources of uncertainty were analyzed and evaluated, while the components of uncertainty were composed. The sources of uncertainty were comprehensively analyzed and calculated. The results show that the main sources of uncertainty in the determination process are the preparation of standard solution and the fitting of standard curve. The content of ethyl maltol in vegetable oil was 95.6 μg/kg, and the expanded uncertainty is 4.3 μg/kg (k=2, 95% confidence interval). This method accurately reflects the confidence and accuracy of the measurement, which can provide technical references for the daily actual inspection work.
Keywords: ultra-high performance liquid chromatography-tandem mass spectrometry; edible vegetable oil; ethyl maltol; uncertainty
乙基麥芽酚因其具有特殊香味,是一種允許使用的食品用合成香料,作為香味增效劑和香味改良劑被廣泛用于煙草、食品、飲料、香料和日用化妝品等行業(yè)[1]。乙基麥芽酚具有還原性,可作為抗氧化劑使用,能與金屬形成贅合物,可控制人體內(nèi)鐵和鋁的含量水平[2-3]。但《食品安全國家標(biāo)準(zhǔn) 食品添加劑使用標(biāo)準(zhǔn)》(GB 2760—2014)附錄B中明確規(guī)定植物油脂中不得添加食品用香料、香精。一些不法商家為牟取利益,在食用植物油中添加乙基麥芽酚,以次充好。為保障人體健康和安全,需準(zhǔn)確檢測食用植物油中乙基麥芽酚含量。
目前,乙基麥芽酚的檢測方法主要有氣相色譜法[4-5]、液相色譜法[6-7]、液相色譜-串聯(lián)質(zhì)譜法[8-9]和拉曼光譜法[10]。有關(guān)使用液相色譜-串聯(lián)質(zhì)譜法測定植物油中乙基麥芽酚的不確定度判定報道不多,測量不確定度是對測量結(jié)果質(zhì)量的定量表征,測量結(jié)果的準(zhǔn)確性很大程度上取決于其不確定度的大小,建立各種檢測的不確定度評定方法,既是提高檢測質(zhì)量的要求,也是實(shí)現(xiàn)檢測數(shù)據(jù)國際互認(rèn)不可或缺的內(nèi)容[11-12]。研究依據(jù)國家市場監(jiān)督管理總局發(fā)布的食品補(bǔ)充檢驗(yàn)方法《食用植物油中乙基麥芽酚的測定》(BJS 201708)進(jìn)行實(shí)驗(yàn),參照現(xiàn)有化學(xué)分析中不確定度評定方法和要求,對超高效液相色譜-串聯(lián)質(zhì)譜法測定食用植物油中乙基麥芽酚含量的不確定度進(jìn)行分析評定。根據(jù)建立的數(shù)學(xué)模型,對影響結(jié)果的相關(guān)分量進(jìn)行不確定度評估,計(jì)算合成相對不確定度,得到最終的擴(kuò)展不確定度,以期為測量結(jié)果的準(zhǔn)確性提供科學(xué)依據(jù)。
1 材料與方法
1.1 材料與試劑
乙腈、甲醇(均為色譜純,德國Merck公司);甲酸(色譜純,上海安普實(shí)驗(yàn)科技股份有限公司);乙酸銨(優(yōu)級純,山東西亞化工科技有限公司);乙基麥芽酚(純度>99%,上海安譜實(shí)驗(yàn)科技股份有限公司)。
1.2 儀器與設(shè)備
Agilent 1290超高效液相色譜儀;Agilent 6470 QQQ三重串聯(lián)四級桿質(zhì)譜;LT2002電子天平,常熟市天量儀器有限公司;UMV-2多管渦旋混合器,北京普立泰科儀器有限公司;Milli-Q超純水機(jī),美國Millipore公司;0.22 μm有機(jī)濾膜,上海安譜實(shí)驗(yàn)科技股份有限公司。
1.3 實(shí)驗(yàn)方法
1.3.1 標(biāo)準(zhǔn)溶液配制
①標(biāo)準(zhǔn)儲備溶液配制。準(zhǔn)確稱取乙基麥芽酚標(biāo)準(zhǔn)品101 mg(精確至0.000 1 g),用甲醇溶解并定容至100 mL,得到濃度為1.0 mg/mL標(biāo)準(zhǔn)儲備溶液。②標(biāo)準(zhǔn)中間溶液配制。吸取標(biāo)準(zhǔn)儲備溶液0.40 mL于100 mL容量瓶中,用甲醇稀釋并定容至刻度,得到
4.0 μg/mL標(biāo)準(zhǔn)中間溶液。
1.3.2 儀器條件
(1)液相色譜條件。色譜柱:ZORBAX Eclipse Plus-C18(50 mm×2.1 mm,1.8 μm);流動相A:0.1%甲酸水溶液,流動相B:乙腈;流速:0.3 mL/min;柱溫:40 ℃;進(jìn)樣體積:2.0 μL;梯度洗脫程序:0~0.5min,5% B;0.5~2.0 min,5%→90% B;2.0~2.5 min,90% B;2.5~2.8 min,90%→5% B;2.8~3.5 min,5% B。
(2)質(zhì)譜條件。離子源:電噴霧離子源,正離子掃描;多反應(yīng)監(jiān)測;毛細(xì)管電壓:4.5 kV;霧化器壓力:40 psi;干燥氣流速:10 L/min;干燥氣溫度:300 ℃;鞘氣流速:10 L/min;鞘氣溫度:300 ℃。其他質(zhì)譜條件見表1。
1.3.3 樣品前處理
準(zhǔn)確稱取10 g試樣(精確至0.01 g)置于50 mL聚丙烯離心管中,用移液器準(zhǔn)確加入10 mL甲醇,渦旋振搖2 min,4 ℃條件下9 000 r/min離心10 min,將上清液移入20 mL具塞刻度試管中,下層油液再用10 mL甲醇重復(fù)提取一次,合并上清液,用甲醇定容至20 mL,經(jīng)微孔濾膜(0.22 μm有機(jī)相)過濾,供液相色譜-串聯(lián)質(zhì)譜分析。
1.3.4 建立不確定度數(shù)學(xué)模型
不確定度數(shù)學(xué)模型如下:
(1)
式中:X為試樣中乙基麥芽酚的含量,μg/kg;C為由工作曲線得出的試樣溶液中乙基麥芽酚的濃度,ng/mL;V為試樣溶液定容體積,mL;m為試樣質(zhì)量,g;為加標(biāo)回收率修正因子。
2 結(jié)果與分析
2.1 不確定度來源
從測量過程和數(shù)學(xué)模型分析,樣品測定的不確定度來源主要有標(biāo)準(zhǔn)溶液的配制、稀釋urel(Std)、標(biāo)準(zhǔn)曲線的擬合urel(C)、樣品前處理urel(Q)、測量重復(fù)性urel(X)和加標(biāo)回收率urel(R)等。
2.2 標(biāo)準(zhǔn)溶液配制、稀釋引入的不確定度urel(Std)
2.2.1 標(biāo)準(zhǔn)品純度引入的不確定度urel(P)
查標(biāo)準(zhǔn)物質(zhì)證書,乙基麥芽酚標(biāo)準(zhǔn)物質(zhì)純度為99.0%,誤差為±0.5%,考慮矩形分布,包含因子k=,屬B類評定,則標(biāo)準(zhǔn)品標(biāo)準(zhǔn)不確定度為;標(biāo)準(zhǔn)品相對標(biāo)準(zhǔn)不確定度為。
2.2.2 標(biāo)準(zhǔn)品稱量引入的不確定度urel(w)
(1)標(biāo)準(zhǔn)品稱量使用萬分之一天平,要求稱量精確至0.000 1 g。天平檢定證書給出的允差為±0.05 mg,考慮矩形分布,包含因子k=,屬B類評定,則標(biāo)準(zhǔn)不確定度 mg;相對標(biāo)準(zhǔn)不確定度。
(2)天平重復(fù)性標(biāo)準(zhǔn)偏差為±0.02 mg,作為標(biāo)準(zhǔn)品稱量重復(fù)性不確定度,則相對標(biāo)準(zhǔn)不確定度。
因此,標(biāo)準(zhǔn)物質(zhì)稱量的合成相對標(biāo)準(zhǔn)不確定度為:
2.2.3 標(biāo)準(zhǔn)儲備溶液配制引入的不確定度urel(C1)
(1)100 mL單標(biāo)線容量瓶(A級)引入的不確定度。標(biāo)準(zhǔn)儲備液配制使用的100 mL單標(biāo)線容量瓶(A級),從《常用玻璃量器檢定規(guī)程》(JJG 196—2006)中可知,100 mL單標(biāo)線容量瓶(A級)允差為±0.10 mL,考慮三角分布,包含因子k=,屬B類評定,則標(biāo)準(zhǔn)不確定度為 mL;相對標(biāo)準(zhǔn)不確定度為。
(2)溫度變化引入的不確定度。實(shí)驗(yàn)室溫度在(20±5)℃變動,甲醇的膨脹系數(shù)(20 ℃)為1.18×10-3/℃,對100 mL容量瓶由溫度效應(yīng)產(chǎn)生的體積變化為ΔV =100×5×1.18×10-3 =0.59,
考慮均勻分布,包含因子k=,屬B類評定,則標(biāo)準(zhǔn)不確定度為 mL;相對標(biāo)準(zhǔn)不確定度為。
(3)量器使用重復(fù)性引入的不確定度。裝滿溶液至100 mL容量瓶刻度10次,稱重并計(jì)算標(biāo)準(zhǔn)偏差為0.02 mL,故重復(fù)性引入的相對不確定度為。
因此,標(biāo)準(zhǔn)儲備溶液配制的合成相對標(biāo)準(zhǔn)不確定度為:
2.2.4 標(biāo)準(zhǔn)中間溶液配制引入的不確定度urel(C2)
(1)1 mL分度吸量管(A級)引入的不確定度。標(biāo)準(zhǔn)中間溶液稀釋過程使用1 mL分度吸量管(A級),從《常用玻璃量器檢定規(guī)程》(JJG 196—2006)中可知,1 mL分度吸量管(A級)容量允差為±0.008 mL,考慮三角分布,包含因子k=,屬B類評定,則標(biāo)準(zhǔn)不確定度為 mL;相對標(biāo)準(zhǔn)不確定度為。
(2)溫度變化引入的不確定度。實(shí)驗(yàn)室溫度在(20±5)℃變動,甲醇的膨脹系數(shù)(20 ℃)為1.18×10-3/℃,對1 mL分度吸量管(A級)由溫度效應(yīng)產(chǎn)生的體積變化為 ΔV=0.4×5×1.18×10-3=2.36×10-3 mL,考慮均勻分布,包含因子k=,屬B類評定,則標(biāo)準(zhǔn)不確定度為 mL;相對標(biāo)準(zhǔn)不確定度為。
(3)100 mL單標(biāo)線容量瓶(A級)引入的相對標(biāo)準(zhǔn)不確定度為。
(4)100 mL單標(biāo)線容量瓶(A級)量器使用重復(fù)性引入的相對標(biāo)準(zhǔn)不確定度為urel(n2)=2.00×10-4。
(5)100 mL單標(biāo)線容量瓶(A級)實(shí)驗(yàn)室溫度變化引起溶劑體積變化引入的相對標(biāo)準(zhǔn)不確定度為。
因此,標(biāo)準(zhǔn)中間溶液配制配制的合成相對標(biāo)準(zhǔn)不確定度為:
2.2.5 標(biāo)準(zhǔn)工作液配制引入的不確定度urel(C3)
實(shí)驗(yàn)中,采用陰性試樣添加標(biāo)準(zhǔn)溶液,分別吸取0.10 mL、0.20 mL、0.40 mL、0.60 mL、0.80 mL和1.00 mL標(biāo)準(zhǔn)中間溶液加入試樣基質(zhì)中,與試樣同時進(jìn)行提取,制成最終濃度為20.0 ng/mL、40.0 ng/mL、80.0 ng/mL、120.0 ng/mL、160.0 ng/mL和200.0 ng/mL標(biāo)準(zhǔn)系列工作液。配制標(biāo)準(zhǔn)工作液過程移取溶液引入的不確定度來源有使用1 mL分度吸量管(A級)、因?qū)嶒?yàn)室溫度變化引起的溶劑體積膨脹等。1 mL分度吸量管(A級)按三角分布,包含因子k=;溫度變化考慮均勻分布,包含因子k=,配制標(biāo)準(zhǔn)工作液引入的不確定度見表2。因此,陰性試樣添加標(biāo)準(zhǔn)溶液1 mL分度吸量管(A級)引入的相對標(biāo)準(zhǔn)不確定度為:
綜上,得出標(biāo)準(zhǔn)溶液配制、稀釋的合成相對標(biāo)準(zhǔn)不確定度為:
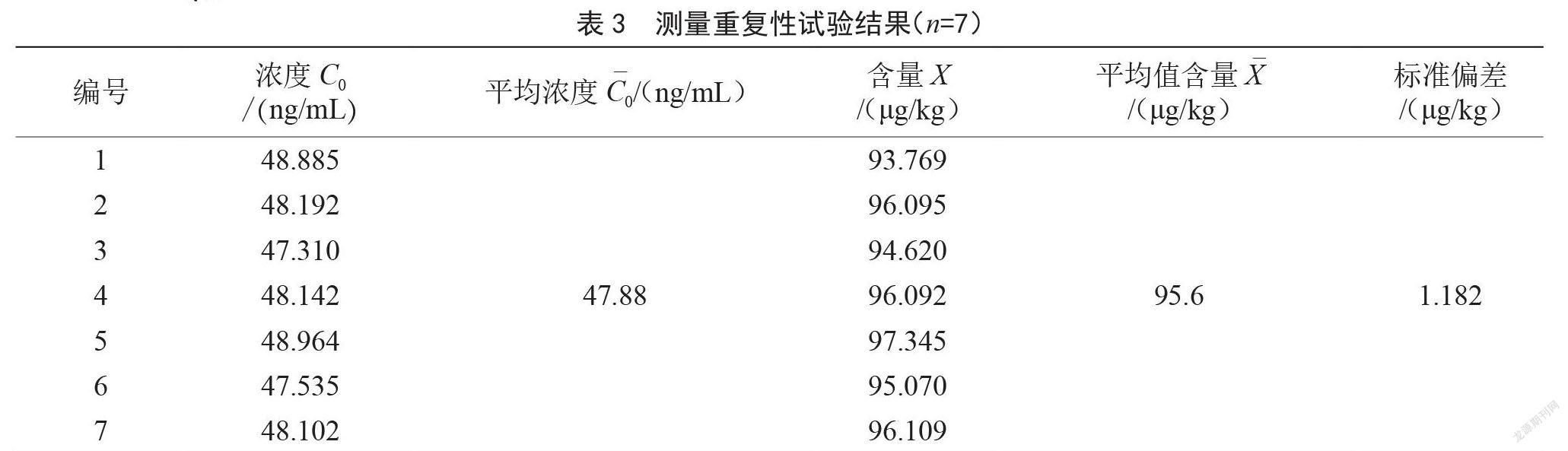
2.3 樣品測量重復(fù)性引入的不確定度urel(X)
通過7次平行試樣檢測,計(jì)算檢測重復(fù)性不確定度,此不確定度結(jié)果已綜合了樣品均勻性、前處理、儀器性能等的不確定度,結(jié)果見表3,按A類評定計(jì)算。重復(fù)性標(biāo)準(zhǔn)偏差 μg/kg。重復(fù)性標(biāo)準(zhǔn)不確定度 μg/kg。重復(fù)性測量相對不確定度。
2.4 標(biāo)準(zhǔn)曲線擬合引入的不確定度urel(C)
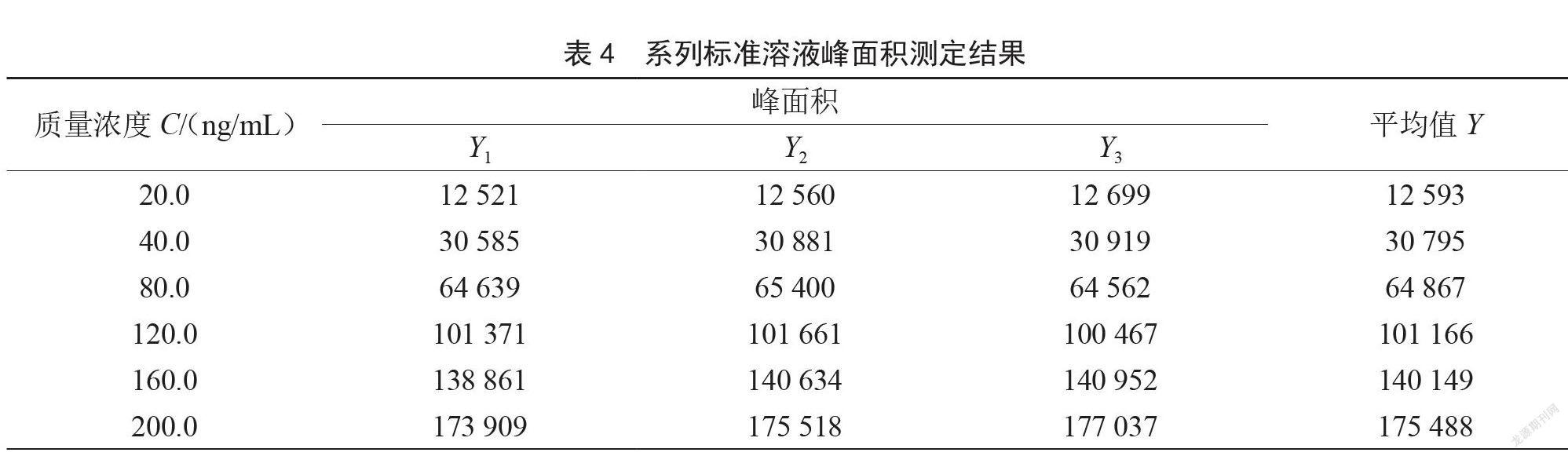
使用液相色譜-串聯(lián)質(zhì)譜儀對6個不同濃度的標(biāo)準(zhǔn)工作液進(jìn)行測定,每個質(zhì)量濃度分別測3次,以質(zhì)量濃度為橫坐標(biāo),峰面積為縱坐標(biāo),通過直線擬合得到標(biāo)準(zhǔn)曲線,線性回歸方程為Y=907.331X-
6 247.866,相關(guān)系數(shù)r=0.999 7,測定結(jié)果見表4,按A類評定計(jì)算。
樣品重復(fù)測定7次,帶入線性回歸方程計(jì)算測定濃度。測得樣品中乙基麥芽酚的平均含量為C0=47.88 ng/mL,各標(biāo)準(zhǔn)溶液質(zhì)量濃度的平均值C=103.33 ng/mL,標(biāo)準(zhǔn)曲線擬合引入的標(biāo)準(zhǔn)不確定度和相對標(biāo)準(zhǔn)不確定度計(jì)算公式如下:
(2)
(3)
式中:S為標(biāo)準(zhǔn)溶液峰面積殘差的標(biāo)準(zhǔn)差;n為樣品平行測定次數(shù),為7;p為標(biāo)準(zhǔn)溶液各濃度點(diǎn)重復(fù)測定的次數(shù),為18;C0為所測樣品溶液的平均質(zhì)量濃度,ng/mL;C表示各標(biāo)準(zhǔn)溶液質(zhì)量濃度點(diǎn)Ci的平均值,ng/mL;k為工作曲線斜率;b為標(biāo)準(zhǔn)曲線截距;Ci為標(biāo)準(zhǔn)溶液各濃度點(diǎn)的含量,ng/mL。
其中,,經(jīng)計(jì)算得到標(biāo)準(zhǔn)曲線擬合引入不確定度u(c)=0.827,相對標(biāo)準(zhǔn)不確定度urel(c)=1.73×10-2。
2.5 樣品前處理引入的不確定度urel(Q)
2.5.1 樣品稱量引入的不確定度urel(m)
稱取植物油10 g試樣(精確至0.001 g)。天平校準(zhǔn)證書給出的允差為±0.005g,考慮矩形分布,包含因子k=,屬B類評定,則標(biāo)準(zhǔn)不確定度 g;相對標(biāo)準(zhǔn)不確定度。
2.5.2 樣品溶液添加引入的不確定度urel(V)
(1)瓶口分液器引入的不確定度。實(shí)驗(yàn)過程中,甲醇提取液的添加使用25.0 mL瓶口分液器。當(dāng)分液器定值為10.0 mL時其容量允差為±50.0 μL,服從三角分布,包含因子k=,屬于B類評定,則標(biāo)準(zhǔn)不確定度 mL;相對標(biāo)準(zhǔn)不確定度。
(2)使用瓶口分液器由溫度變化產(chǎn)生的體積變化 ΔV=10×5×1.18×10-3=0.059 mL,考慮均勻分布,包含因子k=,屬B類評定,則標(biāo)準(zhǔn)不確定度為 mL;相對標(biāo)準(zhǔn)不
確定度為。瓶口分液器使用2次,因此,樣品溶液添加的相對標(biāo)準(zhǔn)不確定度為:
綜上,樣品前處理引入的合成標(biāo)準(zhǔn)不確定度為:
2.6 回收率引入的不確定度urel(R)
取一陰性樣品進(jìn)行加標(biāo)回收試驗(yàn),按1.3.3樣品前處理進(jìn)行處理,平行測定7次,加標(biāo)回收試驗(yàn)結(jié)果見表5。加標(biāo)回收標(biāo)準(zhǔn)不確定度;加標(biāo)回收相對不確定度。為確定回收率是否計(jì)入乙基麥芽酚含量的計(jì)算中,根據(jù)《化學(xué)分析中不確定的評估指南》(CNAS—GL006:2019),對平均回收率R與1.0進(jìn)行顯著性檢驗(yàn),。查t分布表,取置信區(qū)間為95%時,t(0.05,17)>2.447,說明平均回收率R與1.0具有顯著差異性,因此在計(jì)算公式中必須采用回收率校正因子對結(jié)果進(jìn)行修正。
2.7 合成標(biāo)準(zhǔn)不確定度及擴(kuò)展不確定度
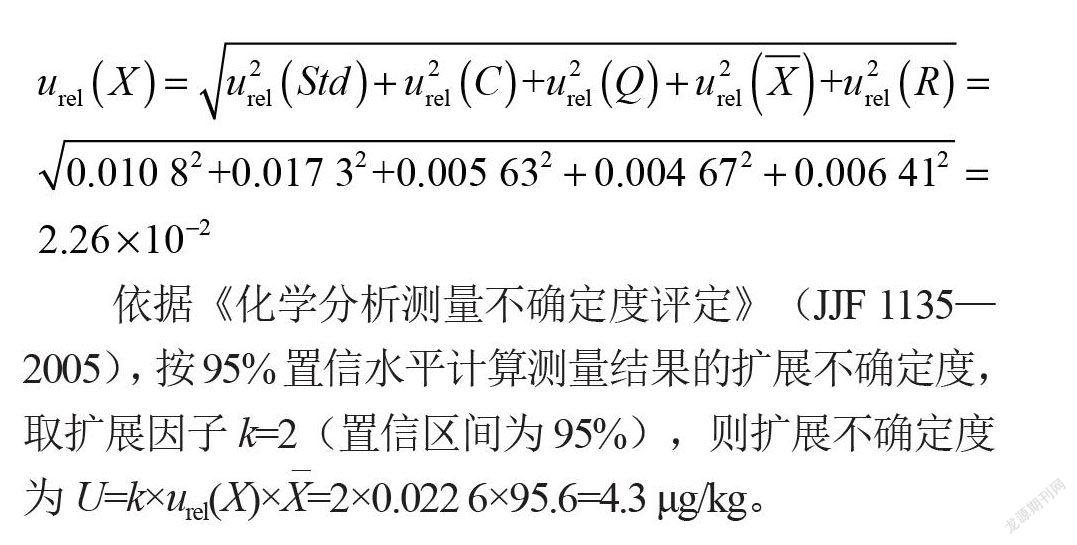
樣品中乙基麥芽酚的測量不確定度各分量見表6。則合成相對標(biāo)準(zhǔn)不確定度為:
依據(jù)《化學(xué)分析測量不確定度評定》(JJF 1135—2005),按95%置信水平計(jì)算測量結(jié)果的擴(kuò)展不確定度,取擴(kuò)展因子k=2(置信區(qū)間為95%),則擴(kuò)展不確定度為u=k×urel(x)×x=2×0.022 6×95.6=4.3 μg/kg。
2.8 測量結(jié)果及不確定度報告
使用超高效液相色譜-串聯(lián)質(zhì)譜法測定植物油中乙基麥芽酚含量,根據(jù)不確定度判定結(jié)果,置信區(qū)間為95%時,最終測定結(jié)果為X=(95.6±4.3)μg/kg,k=2。
3 結(jié)論
采用超高效液相色譜-串聯(lián)質(zhì)譜法測定食用植物油中乙基麥芽酚含量,通過對實(shí)驗(yàn)過程中引入的不確定度分析量化。結(jié)果表明,影響食用植物油中乙基麥芽酚測定結(jié)果不確定度的因素主要為標(biāo)準(zhǔn)溶液配制與稀釋、標(biāo)準(zhǔn)曲線擬合,樣品前處理、測量重復(fù)性和加標(biāo)回收率對不確定度的貢獻(xiàn)相對較小。因此,在實(shí)際檢測過程中要注意操作的規(guī)范性、一致性及平行樣品的測定問題,以減少測量不確定度,保證檢測結(jié)果的準(zhǔn)確度和可信度。
參考文獻(xiàn)
[1]蘇國強(qiáng).乙基麥芽酚生產(chǎn)與應(yīng)用的新進(jìn)展[J].食品科學(xué),1988(1):15-20.
[2]穆旻,鄭福平,孫寶國,等.麥芽酚和乙基麥芽酚的合成及其在食品工業(yè)中的應(yīng)用[J].中國食品學(xué)報,2006,6(1):407-410.
[3]Li Z,LU J L,WU C H,et al.Toxicity Studies of Ethyl Maltol and Iron Complexes in Mice[J].Biomed Research
International,2017,2017:2640619.
[4]景贊,劉超,劉曉碧.氣相色譜法測定液態(tài)乳中麥芽酚和乙基麥芽酚[J].現(xiàn)代食品,2020(5):199-201.
[5]龔國珍,李媛.氣相色譜法測定食用植物油中乙基麥芽酚的含量[J].檢驗(yàn)檢疫學(xué)刊,2019,29(2):136-137.
[6]王浩,劉艷琴,楊紅梅,等.高效液相色譜法測定飲料中乙基麥芽酚的研究[J].食品科技,2006(8):235-236.
[7]賀江,馬丹露.飲料中乙基麥芽酚UPLC檢測方法的建立[J].中國食品添加劑,2018(1):182-186.
[8]馬斐,李鑫,張佳慧,等.液相色譜-串聯(lián)質(zhì)譜法測定芝麻油中乙基麥芽酚[J].食品安全導(dǎo)刊,2021(26):79-81.
[9]寧霄,何歡,金紹明,等.超高效液相色譜-串聯(lián)質(zhì)譜法同時測定食品中麥芽酚、乙基麥芽酚、香蘭素、甲基香蘭素和乙基香蘭素[J].食品安全質(zhì)量檢測學(xué)報,2017,8(7):2555-2562.
[10]吳昊,肖東方,應(yīng)葉,等.基于反應(yīng)體系的表面增強(qiáng)拉曼光譜快速檢測乙基麥芽酚[J].上海師范大學(xué)學(xué)報(自然科學(xué)版),2020,49(2):167-174.
[11]許輝,程剛,周茜,等.禽肉中金剛烷胺殘留量測定的不確定度評定[J].食品研究與開發(fā),2013,34(22):68-71.
[12]LEITO S, MOLDER K,KUNNAPAS A,et al.Uncertainty in liquid chromatographic analysis of pharmaceutical product: Influence of various uncertainty sources[J].Journal of Chromatography A,2006,1121(1):55-63.
