基于機器學習構建皮膚惡性黑色素瘤轉移特征預測模型的研究
2022-06-22 08:05:26王英倫何孜灝黎思穎鄭子豪董博文鄧展程陳秋銳沈晗
廣東藥科大學學報 2022年3期
王英倫,何孜灝,黎思穎,鄭子豪,董博文,鄧展程,陳秋銳,沈晗,2
(1.廣東藥科大學生命科學與生物制藥學院,廣東 廣州 510006;2.廣東省生物技術候選藥物研究重點實驗室,廣東 廣州 510006)
黑色素瘤是來源于黑色素細胞的一類高度惡性腫瘤,多發于皮膚,早期發現并手術切除原發性皮膚黑色素瘤具有較好療效,然而其發病具有隱匿性強、易轉移等特點,多數患者在晚期發生轉移后才被診斷,預后較差[1]。同時,原發黑色素瘤與轉移黑色素瘤在治療手段上也存在一定差異,因此,及時準確地判斷黑色素瘤的轉移特征對臨床治療具有重要的指導意義。近年來,隨著二代高通量測序技術的發展和應用,已積累了大量腫瘤相關的測序數據,其中以TCGA(The Cancer Genome Atlas)數據庫和GEO(Gene Expression Ommibus)數據庫為代表,這些數據信息為腫瘤相關研究提供了極大便利。然而,面對海量的生物信息數據,傳統研究方法已無法適應要求,AI(artificial intelligence,人工智能)技術的發展讓科學家可以借用電腦實現數據的高效快速分析,機器學習是AI 中最具智能特征、最前沿的研究領域之一[2]。本文選取TCGA 數據庫及GEO數據庫中皮膚黑色素瘤的轉錄組數據及臨床特征信息,運用機器學習XGBoost(eXtreme Gradient Boosting)算法,構建皮膚惡性黑色素瘤轉移特征預測模型,旨在為幫助判斷皮膚黑色素瘤患者的臨床分型及分期提供有益的借鑒。
1 方法
1.1 數據下載與整理
從TCGA 數據庫網站(https://portal.gdc.cancer.gov/)下載470 例皮膚惡性黑色素瘤樣本數據,包含原始RNA-Seq Count 數據、RNA 表達FPKM(Fragments Per Kilobase of exon model per Million mapped fragments)數據和臨床信息數據,其中103 例為原發性腫瘤(TP)性狀,367 例為轉移性腫瘤(TM)性狀,將FPKM 數據整理轉換成TPM(Transcripts Per Kilobase of exon model per Million mapped reads)數據。從GEO 數據庫網站(https://www.ncbi.nlm.nih.gov/gds/)下 載GSE190113 數 據集,其包含103 例皮膚惡性黑色素瘤樣本的RNA 表達TPM 數據和臨床信息數據,所有數據分析前進行標準化處理。
1.2 差異表達基因分析
分別利用R 語言(version 4.0.5)中的edgeR 包[3]與DESeq2 包[4],以TCGA 原始RNA-Seq Count 數據制成的基因表達矩陣和對應的臨床信息數據作為輸入資料,進行兩次差異分析,完畢后,將兩者所得結果以P<0.05 且|log2FoldChange|>1 為篩選閾值,獲取表達差異顯著基因。并將兩種算法獲得的差異表達基因取交集。為驗證結果,使用交集差異表達基因的TPM 數據,使用R 語言中的FactoMineR包[5]進行了主成分分析(principal component analysis,PCA)。
1.3 加權基因共表達網絡分析(WGCNA分析)
WGCNA 分析旨在找出協同表達的基因模塊,探索基因網絡與轉移特征表型之間的關聯,從而篩選出相關程度高的基因模塊以及網絡中的核心基因。利用R 語言中的WGCNA 包[6],為加快運算速度同時減少噪音,我們計算了基因表達量在樣本內的方差,選取方差排名前25%的基因制成表達矩陣進行層析聚類,剔除明顯離群樣本。導入篩選過后的基因表達矩陣與轉移相關性狀(TM 與TP),選取恰當的β值作為軟閾值,構建拓撲重疊矩陣,實現無標度網絡。隨后進行動態樹剪切,將相似模塊融合,完成WGCNA分析。
1.4 生物學功能和通路富集分析
利用R 語言中的clusterProfiler 包[7],對轉移特征相關模塊中的基因進行GO 富集分析(生物學進程BP、細胞成分CC、分子功能MF),并利用GSEA軟件[8](version 4.1.0)對全部基因進行GSEA 富集分析驗證GO分析結果。
1.5 ROC分析
利用R 語言中的pROC 包[9],以TP 和TM 為特征,對全部基因進行批量化ROC 分析,繪制每個基因的ROC 曲線并計算該曲線下的面積AUC 值,并篩選AUC>0.7的基因。
1.6 機器學習XGBoost分類模型的構建
對差異分析中的差異表達基因、WGCNA 分析中轉移特征相關模塊內的基因和ROC 分析中AUC值大于0.8 的基因進行交集分析,篩選交集作為訓練模型中使用的特征。利用R語言中的mlr包[10],選擇XGBoost(eXtreme Gradient Boosting,極限梯度boosting)算法并選用上述特征,構建出一個可區分轉移性或非轉移性惡性黑色素瘤的機器學習模型,具體方法如下:將470 個樣本隨機抽樣分為兩部分數據集,4/5 用作訓練集,1/5 用作內部測試集(所有的分割數據集步驟中均確保兩種類型的樣本的抽取比例與原始比例相等);輸入訓練集,使用XG‐Boost 算法進行五折交叉驗證、隨機搜索1000 次的超參數調優獲取最佳超參數組合,以MMCE(mean misclassification error,平均誤分類誤差)作為評估指標;將最優超參數組合應用于XGBoost 算法,并用其來學習訓練集數據,獲得機器學習分類模型;為驗證包括超參數調優步驟在內的整個模型構建過程,評估該算法在訓練集上的性能,采用嵌套交叉驗證,其外循環為五折交叉驗證,內循環為超參數調優過程。至此,獲得以惡性黑色素瘤中基因表達量為特征判別非轉移性或轉移性的模型。最后,使用該模型預測內部測試集中以測試其泛化能力,并進一步選取GEO 數據庫中的GSE190113 數據集作為外部測試集,驗證該模型預測能力。
2 結果
2.1 TP組與TM組差異表達基因篩選
將TCGA 數據庫來源的皮膚惡性黑色素瘤樣本按轉移特征分為原發腫瘤組(TP 組)和轉移腫瘤組(TM 組),使用R 語言的edgeR 包與DESeq2 包進行差異分析,分別獲得2041 個與3846 個差異表達基因,將兩者結果取交集后獲得1804 個差異表達基因。如圖1所示為edgeR包分析火山圖,如圖2所示為DESeq2 包分析火山圖,以TP 組作為對照組,藍色點表示下調基因,紅色點表示上調基因,灰色點則表示表達差異不顯著的基因。交集后差異表達基因的PCA 分析結果顯示TP 組和TM 組存在差異表達現象(圖2)。
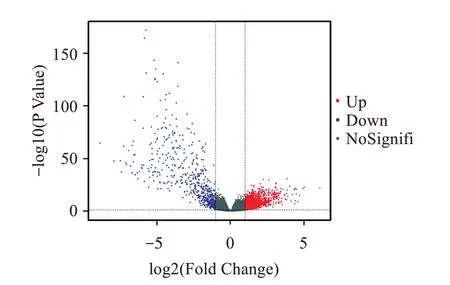
圖1 edgeR包分析差異基因火山圖Figure1 Differential gene volcano plots analyzed with the edgeR package

圖2 DESeq2 R包分析差異基因火山圖Figure 2 Differential gene volcano plots analyzed with the DESeq2 R package
2.2 WGCNA分析
WGCNA 分析結果顯示,發現35 個轉移性狀相關模塊(圖4),其中black 模塊與TM 性狀的負相關性最高,r=-0.46,同時black 模塊與TP 性狀的正相關性最高,r=0.46,該模塊包含529個基因,在TM 組中呈下調表達。
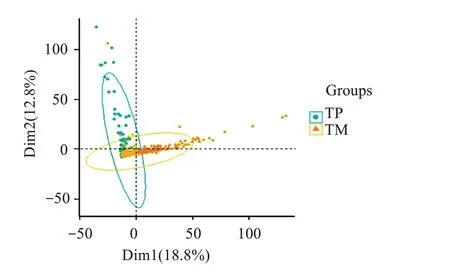
圖3 交集差異表達基因的PCA分析Figure3 PCA analysis of intersection differentially expressed genes
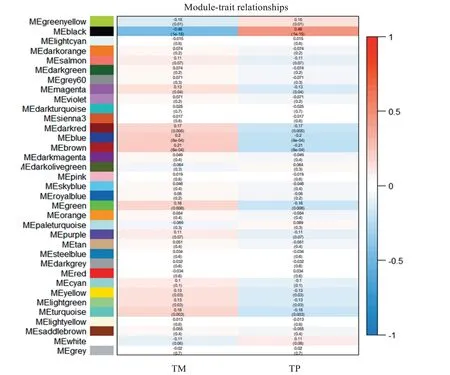
圖4 模塊與轉移性狀的WGCNA分析Figure4 WGCNA analysis of modular and metastatic traits
2.3 GO與GSEA富集分析
GO 富集分析結果提示(圖5),在生物學進程(BP)方面,black 模塊中基因主要參與了表皮發育、分化等進程,在細胞成分(CC)方面,模塊基因與細胞-細胞連接、角質化包膜、中間絲細胞骨架等組分密切相關,在分子功能(MF)方面,模塊基因主要參與了細胞內肽酶、絲氨酸水解酶等酶活性調節及細胞骨架構建等功能。

圖5 black模塊基因的GO富集分析Figure 5 GO enrichment analysis of black module genes
為進一步分析轉移特征基因集的分布情況,對TM 組和TP 組進行了GSEA 富集分析,圖6 結果顯示了在FDRq-val<0.25 條件下,標準化富集得分(NES)最高的10 個功能基因集和NES 最低的10 個功能基因集,圖7 顯示了與TM 組負相關的代表性GSEA 富集詳細信息圖,主要涉及細胞骨架中間絲的構成、黏附以及細胞連接通道活性等生物學過程,提示細胞骨架結構的減弱與腫瘤轉移特征的發生有密切關系。
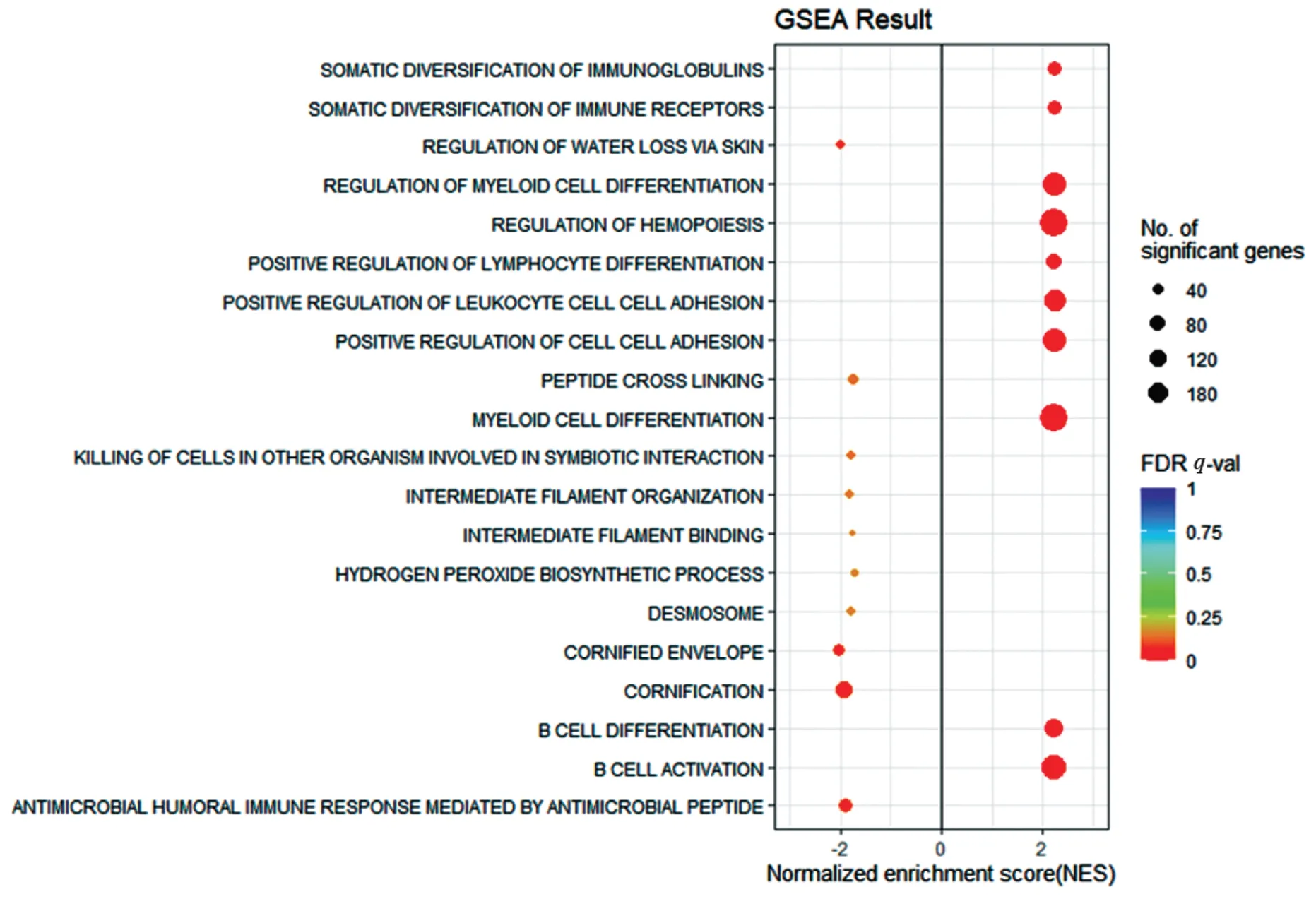
圖6 與TM組相關的GSEA富集分析Figure 6 GSEA enrichment analysis associated with the TM group
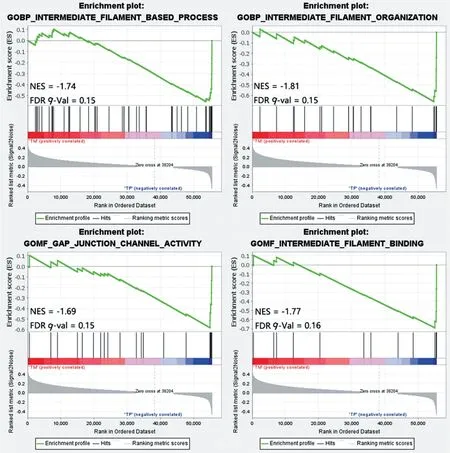
圖7 代表性的標準化GSEA富集評分(NES)與FDR值Figure 7 Representative normalized GSEA enrichment score(NES)vs.FDR values
2.4 ROC分析
基于單個基因的RNA 表達TPM 值,設定TP 與TM為判別特征,進行批量化ROC分析,繪制針對每個基因的ROC 曲線,計算其下面積AUC 值,篩選AUC>0.7的基因,共獲得806個基因,利用此類基因RNA 表達TPM 值對判斷TP 及TM 特征的準確性在70%以上,圖8展示了代表性基因的ROC曲線。

圖8 代表性基因的ROC曲線Figure 8 ROC curves of representative genes
2.5 機器學習XGBoost分類模型的建立及性能評估
基于差異表達基因、black模塊基因和AUC>7.0基因,三者取交集后共獲得143 個基因(圖9),其中AL049555.1基因可能是污染基因,將其排除,剩余142 個基因作為機器學習的特征基因,具體基因名如下:

圖9 三部分分析結果的交集基因維恩圖Figure 9 Venn diagram of intersection genes of three-part analysis results



使用XGBoost 算法構建機器學習模型,為驗證包括超參數調優步驟在內的整個模型構建過程,評估該算法在訓練集上的性能進行的嵌套交叉驗證結果顯示MMCE 值約為0.12,說明該算法在訓練集上表現良好。
使用構建出的分類模型對內部測試集進行預測,內部測試集共有95 例樣本,預測結果繪制混淆矩陣圖[10],如圖10 所示,內部測試集中82 例被正確分類,13例被錯誤分類,MMCE值約為0.14,ROC曲線分析,AUC=0.88。
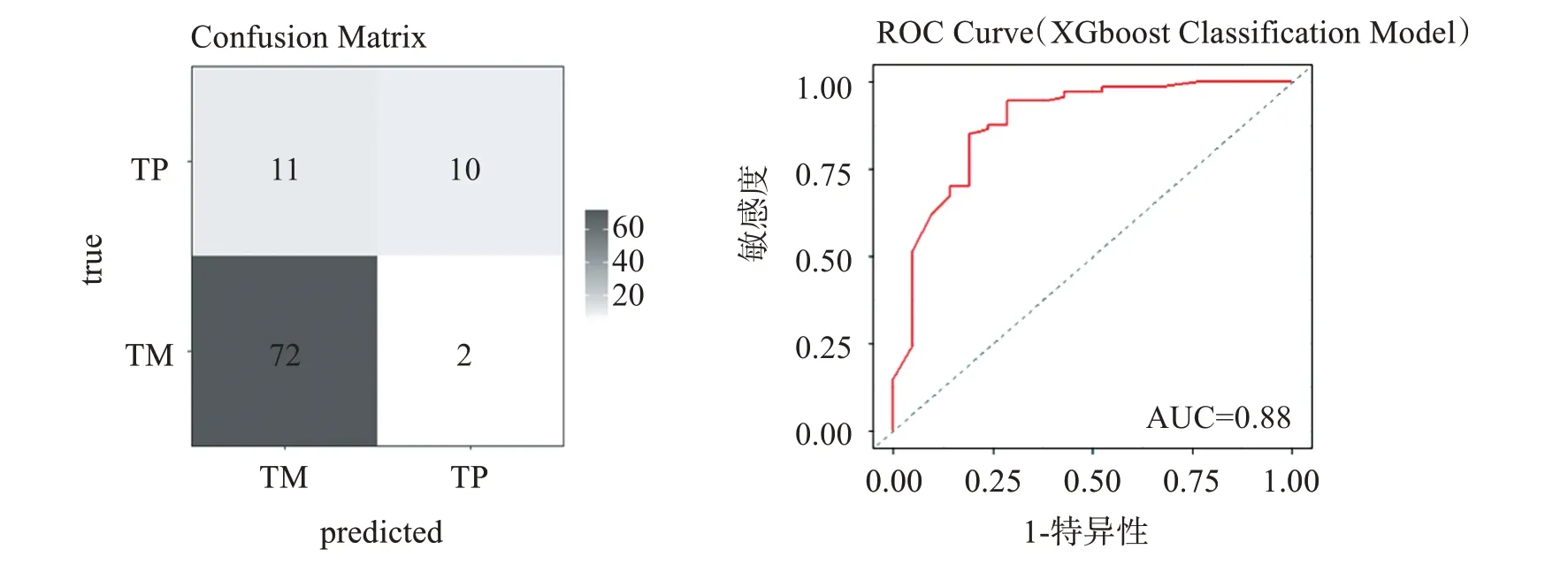
圖10 內部測試集混淆矩陣及ROC曲線Figure 10 Confusion matrix and ROC curve of internal test set
使用構建出的分類模型對外部測試集GSE190113 進行預測,外部測試集共有105 例樣本,如圖11 所示,外部測試集中86 例被正確分類,19 例被錯誤分類,MMCE 值約為0.18,ROC 曲線分析,AUC=0.85。
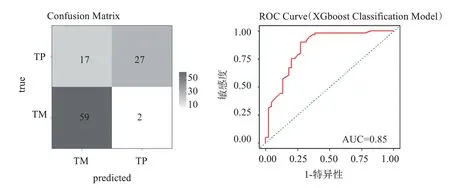
圖11 外部測試集混淆矩陣及ROC曲線Figure 11 Confusion matrix and ROC curve of external test set
3 討論
惡性黑色素瘤占皮膚惡性腫瘤第3位,近年來,其發生率和死亡率逐年升高。由于皮膚惡性黑色素瘤易發生隱匿性轉移,往往轉移后病程進入中晚期,預后較差。因此,及早診斷及早局部手術切除是爭取治愈的最佳方法。目前臨床上,如何有效判斷黑色素瘤的原發灶和轉移灶特征,尚缺乏行之有效的手段。
近年來,隨著生物信息技術的飛速發展,目前已累積了較為可觀的腫瘤相關生物大數據,利用AI技術進行大數據分析利用,在腫瘤輔助診斷[11]、預后模型構建[12]及腫瘤標志物篩查[13]等方面已取得了長足進步。
本研究篩選TCGA 數據庫中,470 例皮膚惡性黑色素瘤樣本,根據其臨床特征分為TP 組和TM組,應用R語言的edgeR包與DESeq2包共篩選出兩組間的差異表達基因1804 個,利用WGCNA 分析篩選出與TM 及TP 性狀相關性最高的black 模塊,該模塊包含529 個轉移特征相關基因,并利用ROC曲線分析獲得806 個AUC>0.7 的轉移特征判別基因,3 種篩選方法取交集,最后確定了142 個轉移特征基因,接著利用XGBoost 算法搭建機器學習模型,以142 個基因的RNA 表達TPM 值為輸入對象,對皮膚惡性黑色素瘤的轉移特征進行預測,通過內部驗證集和外部驗證集評估模型性能,結果證實本模型能有效判斷黑色素瘤的轉移特征,內部測試集的預測準確率為88%,外部驗證集的預測準確率為85%。
本機器學習模型尚存在一些不足之處,首先,由于不同機構采用的檢測方法存在差異,導致轉錄組數據可能存在批次效應,會對模型預測結果產生影響。其次,本模型采用的數據來自TCGA 數據庫,樣本為歐美白人人種,對于我國為代表的東亞人種是否適用尚需檢驗。最后,由于存在數據量和數據特征的局限性,在實際應用中可能會存在偏置性問題。因此,數據的標準化、采用國內患者樣本數據及擴大數據量等措施將是本研究需要進一步改進的方向。
猜你喜歡
音樂探索(2022年2期)2022-05-30 21:01:37
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
瘋狂英語·新策略(2019年10期)2019-12-13 08:43:28
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
當代陜西(2019年10期)2019-06-03 10:12:04
電子制作(2018年18期)2018-11-14 01:48:24
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
數學小靈通·3-4年級(2017年9期)2017-10-13 08:10:54
山東工業技術(2016年15期)2016-12-01 05:31:22
鄭州大學學報(醫學版)(2015年2期)2015-02-27 14:50:46
