慢性粒單核細胞白血病合并單克隆免疫球蛋白血癥1例*
2022-06-27 09:19:00李天壽王健琨黃錦雄
檢驗醫(yī)學與臨床 2022年12期
李天壽,王健琨,劉 琴,黃錦雄
柳州市人民醫(yī)院血液內(nèi)科,廣西柳州 545006
2008年世界衛(wèi)生組織(WHO)將慢性粒單核細胞白血病(CMML)歸入骨髓增生異常綜合征(MDS)/骨髓增殖性腫瘤(MPN)中,MDS/MPN是一組髓系疾病,兼具骨髓發(fā)育異常和骨髓增殖的特點。CMML 患者的臨床表現(xiàn)和預后具有高度異質(zhì)性,目前的治療手段包括去基化治療及異基因造血干細胞移植術。單克隆免疫球蛋白血癥(MGUS)由克隆性B細胞-漿細胞過度分泌單克隆免疫球蛋白所致,常見于漿細胞病及B細胞淋巴瘤。CMML伴發(fā)MGUS的病例國內(nèi)外罕見,現(xiàn)就其報道并進行文獻復習。
1 臨床資料
1.1病歷資料及檢查結果 患者男,64歲,2019年7月因“腹脹”在本科住院,既往身體健康,無發(fā)熱、咳嗽,無頭痛,無牙齦出血、血尿、血便不適。查體:貧血貌,皮膚未見瘀斑及出血點,淺表淋巴結、肝、脾未捫及腫大。入院后查血常規(guī)及白細胞計數(shù)(WBC)手工分類:WBC 72.50×109/L,紅細胞計數(shù)(RBC)3.68×1012/L,血紅蛋白(Hb)72.0 g/L,血小板計數(shù)(PLT)77×109/L,中性粒細胞(N)百分比54%,單核細胞(M)百分比30%,幼稚白細胞百分比6%。骨髓細胞學:骨髓增生明顯活躍,原始粒細胞占8%,原始單核細胞占1.5%,幼稚單核細胞占6%,成熟單核細胞占12.5%,形態(tài)學考慮CMML(圖1)。骨髓活檢病理:骨髓組織增生極度活躍(>90%),粒系核左移,粒紅比例增大,均以中幼及以下階段細胞為主,巨核細胞不少,分葉核為主;偶見胞體小、分葉少的巨核細胞;網(wǎng)狀纖維染色(MF-0級);免疫組化:CD34個別(+)、CD117少許(+)、MPO粒細胞(+),CD3、CD20、CD79a淋巴細胞(+),Lyso散在(+)、TdT(-),形態(tài)學改變不除外MDS/MPN可能(圖2)。骨髓免疫分型:淋巴細胞約占有核細胞的14%,各淋巴亞群分布大致正常,原始區(qū)域細胞約占有核細胞的1%,單核細胞約占有核細胞的35%,表型成熟,粒細胞約占有核細胞的52%,部分考慮存在發(fā)育異常,見圖3。BCR/ABL融合基因陰性;染色體核型分析:46,XY[20]。
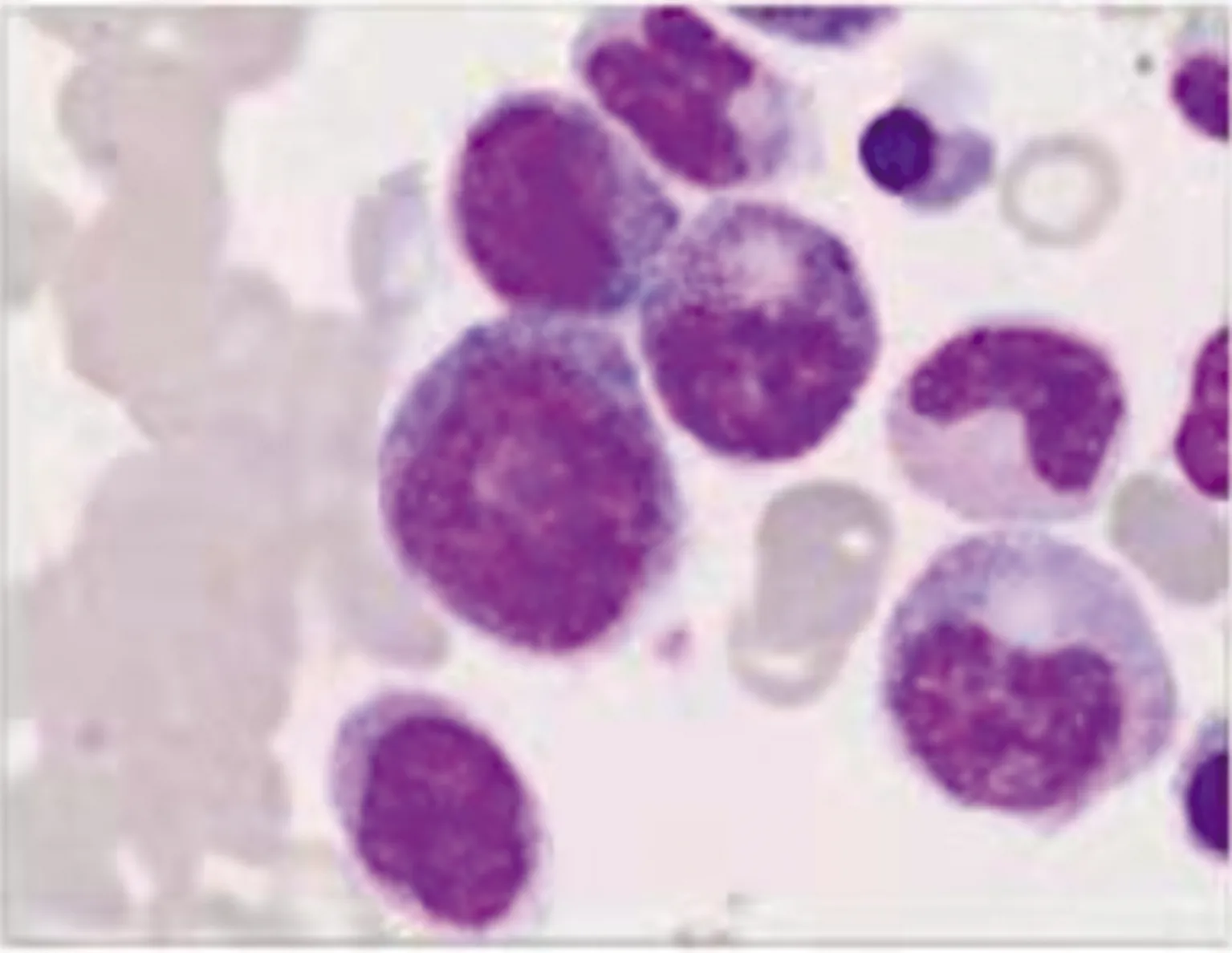
圖1 骨髓細胞形態(tài)學(×1 000)
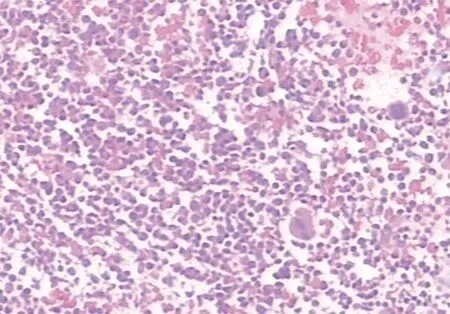
圖2 骨髓活檢病理(×10)
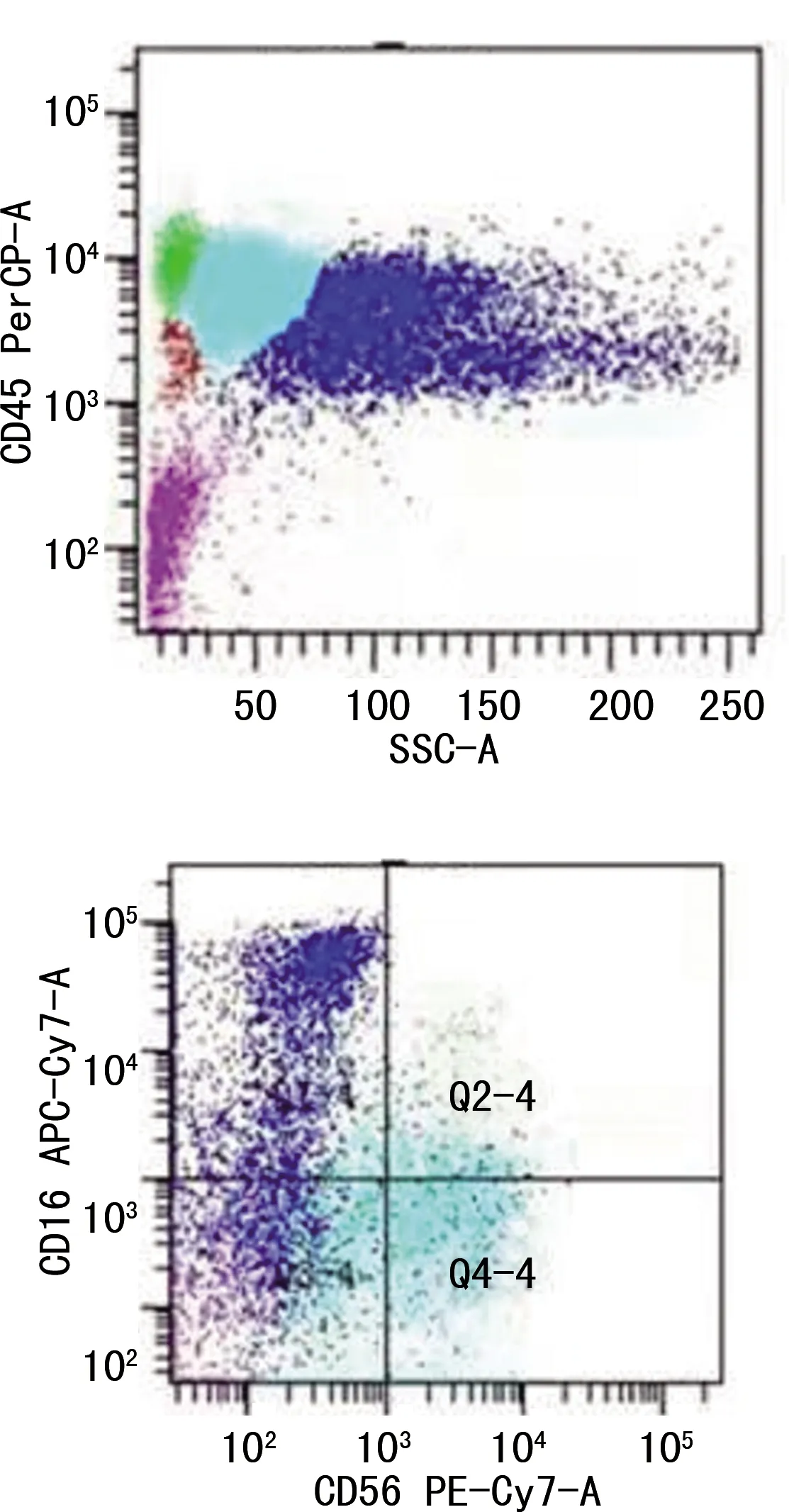
圖3 患者骨髓免疫表型
1.2治療過程 患者分別于2019年8月5日、2019年9月2日接受2個療程地西他濱(20 mg,第1~5天)化療。2019年11月再次返院,查血常規(guī):WBC 43.88×109/L,RBC 3.93×1012/L,Hb 73.0 g/L,PLT 90×109/L,N 2.24×109/L,M 29×109/L。肝功能:總蛋白80.4 g/L,清蛋白30.2 g/L,球蛋白50.2 g/L,IgG 29.7 g/L。血清蛋白電泳:在γ區(qū)見異常濃染條帶,疑為M帶。免疫固定電泳:單克隆免疫球蛋白中IgG陽性,游離輕鏈λ 陽性。β2微球蛋白 5.8 mg/L。骨髓細胞學:原始粒細胞占3.5%,幼稚單核細胞占2.5%。骨髓活檢病理:形態(tài)學改變不除外MDS/MPN可能。骨髓免疫分型:淋巴細胞約占有核細胞的4%,各淋巴亞群分布大致正常;原始及髓系區(qū)域細胞約占有核細胞的83.5%,原始細胞約占有核細胞的1.5%,單核細胞約占有核細胞的40%,部分細胞考慮存在發(fā)育異常,粒細胞約占有核細胞的41%,部分細胞考慮存在發(fā)育異常,另可見約1%的漿細胞,其cKappa/clambda 比值為1.16;提示單核細胞比例明顯升高,伴部分細胞發(fā)育異常。隨后分別于2019年11月23日、2019年12月20日、2020年4月18日、2020年6月6日、2020年10月12日接受地西他濱(20 mg,第1~5天)化療,同時持續(xù)予以口服沙利度胺(100 mg,每晚一次)治療。2020年6月6日返院復查血清蛋白電泳及免疫固定電泳為陰性。隨訪至2021年6月,患者有乏力,無發(fā)熱、出血、骨痛不適,但因經(jīng)濟原因未返院治療。
2 討 論
2008年WHO將CMML歸類為MDS/MPN,這類疾病的臨床和血液學表現(xiàn)兼有MDS和MPN的特點,即骨髓髓系細胞存在過度增殖,而另外的髓系細胞存在病態(tài)造血或無效造血,外周血細胞表現(xiàn)為某類細胞明顯升高,而其他類型細胞減少[1]。該患者外周血WBC、N、M均升高,M大于1×109/L;分子遺傳學檢測未見BCR/ABL融合基因異常,細胞遺傳學正常,排除慢性粒細胞白血病;同時患者外周血及骨髓細胞學提示見幼稚細胞,但原始及幼稚細胞比率<20%;免疫分型提示粒系發(fā)育異常,故患者診斷CMML明確[2]。CMML目前尚無統(tǒng)一治療方案,去甲基化藥物(如地西他濱、阿扎胞苷)聯(lián)合小劑量化療為可能有效的治療方案[3]。國內(nèi)外臨床血液工作者認為異基因造血干細胞移植是唯一能治愈CMML的手段,但移植后仍有一部分患者死于感染或疾病復發(fā)[4-6]。
患者病程中監(jiān)測肝功能提示球蛋白升高,查IgG升高,電解質(zhì)、腎功能正常;影像學檢查未見骨質(zhì)破壞表現(xiàn);進一步完善檢查提示血清蛋白電泳見M帶,免疫固定電泳提示單克隆免疫球蛋白中IgG和游離輕鏈λ陽性;但患者復查骨髓細胞學及免疫分型未見漿細胞明顯異常增生依據(jù);故患者診斷為CMML合并MGUS。MGUS和白血病屬于兩種不同來源的疾病,但白血病合并MGUS并不少見。JIN等[7]報道了1例急性髓系白血病合并MGUS的病例,認為合并MGUS是急性髓系白血病獨立的預后不良因素。意義未明的MGUS屬于漿細胞疾病中的一大類,主要表現(xiàn)為血液中出現(xiàn)大量的單克隆免疫球蛋白,但無異常漿細胞增生及其引起的臨床表現(xiàn)。已有的研究表明,MGUS患者可能會進展為骨髓瘤、華氏巨球蛋白血癥、淀粉樣變性或惡性淋巴瘤[8-9]。關于CMML合并漿細胞疾病的研究較少,楊英等[10]報道了1例CMML合并多發(fā)性骨髓瘤的病例;徐敏等[11]報道了1例冒煙型骨髓瘤合并CMML的病例。這些已報道的病例表明,CMML有可能合并惡性漿細胞疾病,但其中機制尚未明確。本例患者骨髓免疫分析提示異常表達CD56(圖3),CD56 又稱神經(jīng)細胞黏附分子(N-CAM)。研究認為,CD56(+)細胞起源于髓系/自然殺傷細胞的前體細胞,異常表達CD56的血液惡性腫瘤更具侵襲性,預后更差[12]。蔡夢潔等[13]通過研究流式細胞術檢測多發(fā)性骨髓瘤循環(huán)腫瘤漿細胞免疫表型,發(fā)現(xiàn)循環(huán)腫瘤漿細胞CD56陽性率占81.7%。丁雅雯等[14]通過檢測骨髓瘤免疫表型,發(fā)現(xiàn)多發(fā)性骨髓瘤漿細胞CD56陽性率占65.71%。這些研究表明,CD56(+)細胞為更早期的祖細胞,CMML患者CD56(+)腫瘤細胞在腫瘤克隆演化過程中存在漿樣分化可能。
沙利度胺是一種免疫調(diào)節(jié)劑,可抑制腫瘤血管新生,目前較多應用于治療漿細胞疾病及惡性淋巴瘤[15]。SWAMINATHAN等[16]報道了1例慢性粒細胞白血病患者,服用伊馬替尼治療后達到分子學緩解,但停藥后復發(fā),復發(fā)后檢查提示合并MGUS,再次予以伊馬替尼治療后病情得到緩解,隨后檢查發(fā)現(xiàn)合并多發(fā)性骨髓瘤,予以沙利度胺、硼替佐米聯(lián)合地塞米松治療多發(fā)性骨髓瘤,并繼續(xù)予以伊馬替尼治療慢性粒細胞白血病,后患者病情得到控制。也有相關研究證明沙利度胺可應用于治療CMML[17]。KENEALY等[17]開展的一項關于沙利度胺聯(lián)合阿扎胞苷治療MDS、CMML、低增生急性髓系白血病(AML)的臨床研究表明,沙利度胺聯(lián)合阿扎胞苷治療MDS、CMML、AML是安全的,且能提高這3類疾病患者的總生存時間。本例患者病程中發(fā)現(xiàn)合并MGUS,在原有地西他濱的治療方案上加用口服沙利度胺治療。經(jīng)治療,患者乏力癥狀改善,血象WBC得到控制,MGUS轉(zhuǎn)陰。
合并MGUS可能是CMML預后不良的危險因素。CMML合并MGUS罕見,如患者球蛋白升高,且血清蛋白電泳、免疫固定電泳陽性,應完善骨髓形態(tài)學、免疫分型以及遺傳學檢查明確是否合并多發(fā)性骨髓瘤、淋巴瘤可能;地西他濱聯(lián)合沙利度胺治療可能有效。期待有更多CMML合并漿細胞疾病的研究,以闡明其機制。
