膠質母細胞瘤中差異甲基化增強子區域調控的蛋白編碼基因識別研究
2022-06-29 02:25:16趙瀟瀟于秋紅嵇江淮王世佳王仁東李冬果
首都醫科大學學報 2022年1期
趙瀟瀟 于秋紅 嵇江淮 王世佳王仁東李冬果*
(1.首都醫科大學生物醫學工程學院,北京 100069;2.首都醫科大學臨床生物力學基礎研究北京市重點實驗室,北京 100069;3.首都醫科大學附屬北京天壇醫院高壓氧科,北京 100070;4.浙江腫瘤醫院放射物理科,杭州 310022;5.浙江省放射腫瘤學重點實驗室,杭州 310022)
多形性膠質母細胞瘤(glioblastoma,GBM)是最常見且最致命的腫瘤之一,被歸類為Ⅳ級膠質瘤[1]。它是一種高度侵襲性腫瘤其特征是腦血管的改變和周圍組織沿血管空間的逐漸侵襲[2]。GBM細胞通常會侵入距腫瘤塊幾厘米的地方,甚至可以跨入對側半球[3]。目前GBM的治療標準[4]包括手術切除,然后輔以放射治療(以下簡稱放療)和輔助治療。但由于這些腫瘤的彌散性浸潤性質,很少能實現完全切除,GBM患者在治療結束后容易復發,中位生存時間約15個月,且預后較差[5-6]。因此,迫切需要在GBM準確的分子機制和可靠的治療靶點的研究中取得新進展。
癌癥是異常遺傳和表觀遺傳事件的結果。表觀遺傳機制是無法用DNA序列的變化來解釋的穩定遺傳特征,與組蛋白修飾一樣,DNA甲基化并不影響基因組DNA序列本身[7],而是在CG二核苷酸胞嘧啶上增加一個甲基(CH3)基團。在哺乳動物中,5 mC參與了長期的沉默過程,如X染色體失活、基因組印記、生殖系基因的體細胞沉默和重復DNA元素的沉默[8]。根據DNA甲基化的異常變化對腫瘤的診療和預測生物標志物被認為具有廣泛的前景。
近年來,多是對GBM中啟動子異常甲基化動力學進行研究[9],也有少量對增強子甲基化方面的研究[10]。異常甲基化增強子會導致多種疾病,其中包括多種癌癥的基因表達異常[11]。研究[12]顯示,與啟動子相比,增強子區域的甲基化狀態與靶基因表達的相關性更強。然而,只有少量研究[13-14]關注增強子區域且增強子甲基化模式在GBM中仍不清楚。
1 資料與方法
1.1 數據來源及預處理
本研究從基因表達綜合數據庫(gene expression omnibus,GEO)數據庫中下載GBM腫瘤樣本(GSE36278)和正常樣本(GSE42861)的甲基化數據,包括136個腫瘤樣本和58個正常樣本。腫瘤樣本和正常樣本的DNA甲基化數據是通過Infinium HM450k平臺用同樣方法獲得的。從基因組數據共享數據庫(genomic data commons,GDC)數據門戶下載患者臨床數據和136例GBM樣本的表達數據。人類全基因組的注釋數據(V19)收集于GENCODE數據庫[15]。
對于甲基化數據,每個探針的甲基化水平用β值表示。β值=Imeth/(Imeth+Iunmeth),其中Imeth為甲基化強度,Iunmeth為未甲基化強度。為確保甲基化水平的準確性,移除在所有樣本中的甲基化水平超過30%為缺失值的探針。然后,本研究使用 “DMwR” R包中的knnImputation函數[16]對甲基化數據缺失值進行處理。將下載的RNA-seq(FPKM)數據使用log2轉換進行標準化,表達值為0的用數據集中的最小正數代替,最終獲得蛋白編碼基因(protein-coding genes,PCGs)的表達譜數據。
1.2 構建增強子區域和啟動子區域
本研究整合已文獻[17]和GPL13534注釋文件,從而獲得了位于增強子的探針。本研究將這兩部分探針合并,并刪除重復的探針。結果保留161 708個增強子探針用于后續分析。已有研究[18-19]顯示,典型增強子區域的中位間隔大小約為1 000 bp。因此,本研究以一個增強子探針坐標為基準,分別向其上游和下游擴展500 bp的區間來構建增強子區域。重疊的增強子區域被連接,并擴展成較大的區間,然后計算增強子區域中CpG探針的平均值,作為增強子區域的DNA甲基化水平[20]。
由于基因不僅受增強子甲基化的調控,也受啟動子甲基化的調控。為了獲得只受差異甲基化增強子區域(differential methylation enhancer regions, DMERs)調控的基因,本研究需要重新注釋啟動子區域,鑒別出可能受啟動子甲基化調控的基因,然后移除這些基因。本研究定義將基因轉錄起始位點(transcription start site, TSS)上游2 kb區域作為啟動子區,基因的DNA甲基化水平為映射到其啟動子區域的探針平均值[21-22]。
1.3 識別受差異甲基化增強子區域調控的靶基因
本研究利用基于線性模型設計的R包“limma”[23]來識別腫瘤與正常樣本之間的DMERs和差異啟動子甲基化基因(differential promoter methylation genes, DPMGs)。采用Benjamini-Hochberg(BH)方法[24]對P進行校正。同樣,本研究也鑒定了DMERs以進行進一步分析。本研究選擇log2(FC)≥0.2和adjustedP≤0.05 的增強子區域作為高甲基化增強子區域,將log2(FC)≤-0.2 和adjustedP≤0.05 的增強子區域作為低甲基化增強子區域。為了獲得僅受DMERs調控的基因,本研究需要重新注釋啟動子區域,移除可能受啟動子甲基化調控的基因。研究[25]顯示,增強子和基因之間已知的最大距離約為1 Mbp。因此,本研究選擇了位于同一染色體上的DMER-PCGs對,基因的TSS與增強子區域之間的最大線性距離為1 Mbp。本研究用皮爾森相關系數(Pearson correlation coefficient, PCC)計算DMER與基因表達之間的相關性。基因表達被增強子甲基化負調控,高甲基化增強子區域可下調甚至沉默基因表達,而低甲基化增強子區域則傾向于激活基因表達[26],因此本研究只保留了PCC<-0.2的DMER-PCGs對,且P≤0.01。
1.4 功能富集分析
為了預測DMERs調控的PCGs的功能,采用富集分析方法,對受低甲基化增強子區域調控的PCGs進行功能和通路的顯著性分析,使用R包“clusterProfiler”[27]來預測PCGs的功能。通過BH方法校正P,如果校正后的P≤0.05,該生物過程(biological process, BP)和京都基因與基因組百科全書(Kyoto Encyclopedia of Genes and Genomes, KEGG)就認為是顯著的。通過R包‘ggplot2 ’進行顯示。
1.5 生存分析
為了鑒別挑選出的PCGs是否具有良好的預后效果,本研究基于152個GBM患者的表達譜信息,依據中值將患者分為兩組。所有的研究均使用R 4.0.2完成。Kaplan-Meier生存分析和log-rank檢測被用來評估兩組患者的生存差異,以P<0.05為差異有統計學意義。
2 結果
2.1 GBM中增強子區域甲基化譜的構建
為了研究在GBM中增強子的DNA甲基化模式,本研究應用計算策略將增強子探針注釋到Infinium 450K陣列,從而構建GBM增強子區域甲基化譜。在本研究中,增強子探針的收集來自先前發表的文獻的[17]補充文件和GPL13534注釋文件。利用上述方法構造了113 178個非重疊增強子區域。其中增強子區域長度為1 000 bp占比79.99%,長度為>1 000 bp和<2 000 bp占比17.79%,其他占比2.22%(圖1A)。同樣,本研究將Infinium 450K陣列重新注釋到基因的啟動子區域。最終54 477個探針位于20 386個基因啟動子區域。雖然每個基因都有幾個探針定位到相應的啟動子區域,但本研究只計算該基因啟動子中DNA甲基化探針的平均值作為該基因的DNA甲基化水平。
2.2 識別受差異增強子區域調控的基因
在對增強子區域甲基化譜進行預處理后,本研究從136個GBM樣本和58個正常組織樣本中識別出16 287個DMERs,其中包括2 271個高甲基化增強子區域和14 016個低甲基化的增強子區域。類似地,本研究基于基因啟動子甲基化譜共鑒別出4 889個DPMGs。為了獲得表達嚴格受差異啟動子甲基化調控的基因,本研究進一步計算每個DPMG甲基化值與其對應基因表達值之間的PCC。結果顯示,共識別出167個可能受啟動子甲基化調控的基因,其中有144個低甲基化基因和23個高甲基化基因,這些基因將會被排除在研究之外。為了確定哪些基因受DMERs調控,本研究結合多組學數據建立增強子與其靶基因(DMERs-相關基因)的關聯模型(如上方法描述)。
由于已知增強子甲基化水平與染色質活性呈負相關[28],本研究只保留了PCC<-0.2的DMER-基因對。最后,本研究得到了795對DMER-PCGs,其中包含有593個低甲基化增強子區域,82個高甲基化增強子區域和642個PCGs。整體來看,GBM中存在明顯的低甲基化模式(圖1B)。
2.3 靶基因的功能富集分析
為了評估DMERs調控的靶基因的生物學特性,本研究對642個PCGs進行功能富集分析。結果顯示,PCGs富集在許多與腫瘤進程相關的生物過程,比如細胞凋亡、突觸信號傳送與調節、組蛋白H3-K79甲基化的調控(圖1C)。對于KEGG通路富集分析,本研究發現 PCGs在 KEGG中富集出多條與 GBM 相關的通路,比如ErbB(EGFR)、神經營養因子、黏著斑黏附(圖1D)。
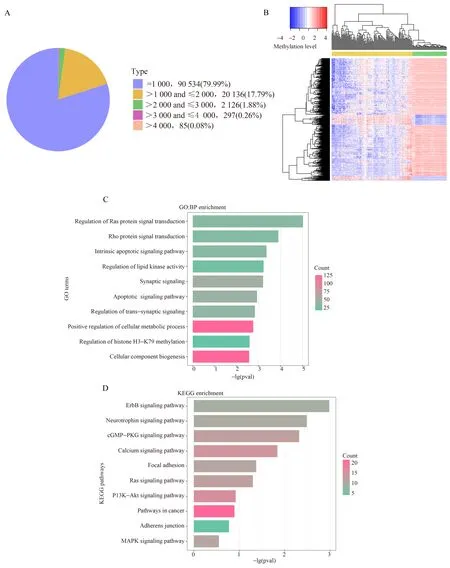
圖1 GBM中增強子區域的DNA甲基化模式
2.4 GBM中預后良好的PCGs的鑒別
為了評估這些表達受DMERs影響的PCGs是否可以作為GBM的預后因子,本研究將表達數據與患者臨床信息相結合,基于生存分析的結果,獲得了45個與GBM整體存活相關的PCGs(P<0.05,表1)。這些基因的異常表達可能是由于增強子異常甲基化調控的。它們可能是GBM潛在的診斷和治療的生物標志物。在本研究中,DTX1和SLC2A3 在GBM中均屬于表達上調的基因。DTX1和SLC2A3 基因高表達組患者的生存期顯著縮短且預后差,可作為GBM患者的獨立預后因素(圖2)。

圖2 DTX1和SLC2A3基因生存分析

表1 預后良好的PCGs
3 討論
研究[12, 29]表明增強子通過甲基化介導表觀遺傳調控,對靶基因的表達進行異常調節,從而導致人類疾病。表觀遺傳調控已成為DNA甲基化等生物醫學研究的熱點[30]。癌癥中DNA甲基化的變化被認為是開發有效的診斷、預后和預測標志物的靶點。最近的研究[31-33]也報道了一些基因啟動子在GBM中的異常甲基化。然而,到目前為止,使用異常甲基化增強子識別GBM生物標志物的研究很少。
本研究通過構建增強子區域,對其進行差異甲基化分析并鑒別出可能受DMERs調控的PCGs,結果發現DMERs在GBM中多呈現一種低甲基化的模式。為了評估PCGs的生物學功能,對這些基因進行功能富集分析。本研究結果顯示PCGs參與了許多與腫瘤發生及進程相關的生物過程。由此推斷這些受DMERs調控的PCGs更有可能成為GBM潛在的診斷和治療的生物標志物。其中EGFR 在GBM 和膠質瘤中所發揮的作用已被廣泛研究,其可以通過促進血管生成和細胞侵襲從而促進腫瘤的惡化[34]。Neurotrophin 為 GBM 發展提供了微環境,并且促進腫瘤浸潤細胞的增生[35]。Huber等[36]發現DTX1的過表達增加了細胞的遷移和侵襲,這與ERK激活、miR-21水平和內源性Notch水平相關。DTX1的Notch通路似乎是膠質母細胞瘤的一種致癌因子,這些發現提供了新的潛在的治療靶點。研究[37-38]表明SLC2A3 在GBM中高表達,沉默期的表達可以抑制GBM細胞的增生和侵襲能力。本研究結果表明DTX1和SLC2A3 在GBM中表達的上調可能是受增強子區域的低甲基化調控,從而導致疾病的發生與發展。
然而,這項研究有一些需要強調的局限性。GBM的樣本采集是一個問題,這一缺陷可能會在一定程度上影響最終的結果。此外,本研究結果基于生物信息學分析。由于技術和時間限制,本研究沒有驗證在GBM動物模型和GBM患者腦組織中的結果。未來的功能研究和分子實驗仍需進一步探索其作用機制。
綜上,本研究通過構建GBM中較為全面的增強子區域的甲基化譜,鑒定出GBM中表達可能受DMERs調控的PCGs,挖掘出與GBM預后相關的PCGs并加深對GBM中差異甲基化增強子調控模式的理解。同時,本研究為在表觀遺傳方面識別潛在的GBM生物標志物和治療靶點提供了新的思路。
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
今日農業(2021年9期)2021-11-26 07:41:24
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
發明與創新·小學生(2021年3期)2021-03-25 11:48:49
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
汽車工程學報(2017年2期)2017-07-05 08:13:02
中國科技博覽(2016年2期)2016-04-25 20:32:39
小學生導刊(2016年34期)2016-04-11 00:49:44
