改性海藻酸鈉-聚乙烯亞胺-氧化石墨烯復合水凝膠對環丙沙星的吸附研究
2022-06-29 04:15:34李茂林戴友芝于芹芹
環境科技 2022年3期
李茂林,戴友芝,張 柱,于芹芹
(湘潭大學 環境與資源學院,湖南 湘潭 411105)
0 引言
環丙沙星(CIP)作為氟喹諾酮類抗生素(FQNs)的代表藥物之一,被廣泛應用于人和動物疾病的治療,但因其易誘導細菌產生耐藥性且對人的心臟和神經系統等造成不良影響,在自然界富集將對生態系統穩定和人類健康構成很大威脅[1]。 對此,常采用吸附法、化學法、膜分離法對環丙沙星進行降解。 其中吸附法是利用多孔性固體吸附劑將液相中的組成吸附于表面,從而達到分離和富集污染物的一種處理方法,因其操作簡便、成本低、污染小、可循環而備受研究者關注[2]。常見的吸附材料包括炭(碳)基材料[3]、高分子材料、新型樹脂等。
海藻酸鈉(SA)作為可生物降解的天然鈉鹽,具有可持續性生物材料基體的特點[4]。其分子主鏈上廣泛分布的羧基官能團可與聚乙烯亞胺(PEI)分子鏈上的胺(-NH2)官能團發生酰胺化反應從而將PEI 負載到SA 上,增強其吸附性能[5]。 氧化石墨烯(GO)是一種比表面積高、富含羥基、羧基、環氧基等含氧官能團的納米材料,其富含的官能團可通過氫鍵作用和Π-Π 作用高效去除抗生素[6]。 但SA-PEI 機械性能弱、易溶于水、穩定性差且對CIP 的吸附能力差。GO 分散于水中回收困難,容易造成二次污染。 水凝膠生物相容性好、結構獨特,在水中不溶解[7]。為彌補SA-PEI 材料的不足和解決GO 回收困難的難題,將GO 引入SA-PEI 體系并通過SA 與Ca2+交聯構筑復合水凝膠[7],從而增加了SA-PEI 的機械強度、比表面積及吸附活性點位。 但由于GO 片層的摻入使凝膠結構更為緊湊,阻礙了水分子的傳輸通道[8],CIP難以進入水凝膠內部,故將碳酸鈣粉末摻入溶膠-凝膠體系后,可增大凝膠球的表面孔徑[9],通過碳酸鈣改性的方式增強海藻酸鈉-聚乙烯亞胺-氧化石墨烯(SA-PEI-GO)凝膠球對CIP 的吸附能力。
1 實驗材料與方法
1.1 主要試劑和儀器
主要試劑:海藻酸鈉、聚乙烯亞胺、氧化石墨、1-乙基-(3-二甲基氨基丙基)碳二亞胺鹽酸鹽、N-羥基琥珀酰亞胺、碳酸鈣、無水氯化鈣、鹽酸、環丙沙星(以上均為分析純)、去離子水。
主要儀器:日本津島AUY 電子分析天平;JJ-1型定時電動攪拌器;KM5200DB 超聲波清洗器;HJ-4A 恒溫多頭磁力攪拌器;pHS-3EpH 計;THZ-82A氣浴恒溫振蕩箱;DZ-2BCIV 真空干燥箱;UV521 紫外分光光度計;SU5000 場發射掃描電子顯微鏡;NICOLET 380 傅里葉紅外光譜儀;NOVA-2200e 物理吸附儀;D/MAX-2500/PCX 射線多晶粉末衍射儀。
1.2 材料制備
采用溶膠-凝膠法制備改性SA-PEI-GO 復合水凝膠。 稱取一定量海藻酸鈉溶于去離子水中后加入EDC·HCl 和NHS,機械攪拌均勻后再按m(SA)∶m(PEI)=10 ∶7 加入聚乙烯亞胺,室溫下繼續攪拌24 h后得到均相水溶膠。同時,稱取少量氧化石墨分散于50 mL 去離子水中,磁力攪拌過夜,形成懸浮液后經超聲處理4 h 即得到GO。 將GO 加入到混合液中機械攪拌混合均勻后,定量稱取碳酸鈣粉末加入其中繼續攪拌均勻,在磁力攪拌條件下,將混合液用分液漏斗和膠頭滴管組裝裝置逐滴滴入濃度為1 mol/L的CaCl2溶液中制得凝膠球。 采用傾析法分離出凝膠球,在磁力攪拌下,將其浸泡于濃度為1 mol/L 的鹽酸溶液中反應12 h 以去除水凝膠中的碳酸鈣。 用去離子水洗滌6 次,再將水凝膠置于去離子水中保存備用。
1.3 表征方法
場發射掃描電鏡及能量散射光譜(SEM-EDS);傅里葉紅外光譜(FT-IR)。
2 吸附實驗
分別稱取1 g(濕重)SA-PEI-GO 和改性SA-PEIGO 復合水凝膠(對應干重均為65 mg)加入到適當的環丙沙星溶液(初始質量濃度為5 mg/L,體積為100 mL)中,在pH 值為7,溫度為(25±0.1)℃,轉速為140 r/min 的氣浴恒溫振蕩箱內振蕩吸附,每隔一段時間分別取1 mL 上清液,經0.22 μm 聚醚砜水系濾頭過濾并稀釋10 倍,采用紫外分光光度計測定吸附后CIP 濃度,吸附容量(Qt)和去除率(R)計算公式如下:

式中:Qt和Qe分別為t 時刻和平衡時的吸附容量,mg/g;C0,Ct和Ce分別為溶液中CIP 在初始時間、t時刻和平衡時的質量濃度,mg/L;V 為水溶液體積,L;m 為吸附劑質量,g。
3 結果分析
3.1 SEM-EDS 表征
未改性和改性凝膠球的SEM 圖像及其吸附CIP 后的EDS 圖譜見圖1。 未改性和改性凝膠球結構中各元素組分占比見表1 和表2。

表1 SA-PEI-GO 中各元素組分占比
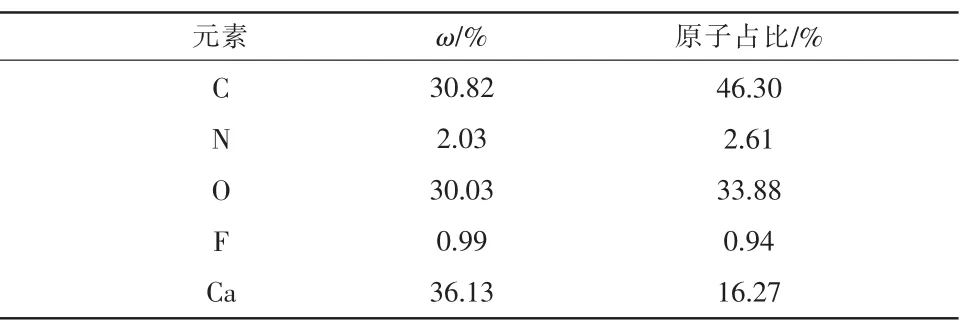
表2 改性后SA-PEI-GO中各元素組分占比


圖1 SA-PEI-GO 和改性SA-PEI-GO 的SEM 圖像及改性SA-PEI-GO 吸附前、后的EDS 圖像
由圖1(a)和圖1(c)可知,凝膠球為近似球形的不規則珠狀材料, 且該樣品因真空干燥脫水而發生了不同程度地收縮。 由圖1(b)和圖1(d)可見,改性SA-PEI-GO 表面形成了更多的褶皺、 塌陷和孔隙,這不僅增加了凝膠球的比表面積,更重要的是打開了CIP 分子進入材料內部及在內部擴散的通道。
由表1和表2可知,C,N,O,Ca 是改性SAPEI-GO 中主要元素,F 元素作為CIP 分子的特征元素出現在吸附后的樣品中,說明CIP 分子被成功吸附至改性SA-PEI-GO 凝膠球上。
3.2 FT-IR 表征
SA,GO,SA-PEI,SA-PEI-GO及改性后SAPEI-GO 的紅外圖譜見圖2。

圖2 SA,GO,SA-PEI,SA-PEI-GO 及改性SA-PEI-GO的紅外圖譜
由圖2 可知,在波數為4 000~1 000 cm-1的范圍內,曲線①和②分別是SA,GO 的紅外圖譜;曲線③是SA-PEI 的紅外圖譜,在波數為3 431cm-1處的強峰是N-H/O-H 的伸縮振動峰,在波數分別為1 631和1 422 cm-1處出現的峰分別為C=O 和C-O 峰,這些吸收峰證明出現了酰胺基團,即PEI 成功負載在SA 上;曲線④是SA-PEI-GO 的紅外圖譜,在加入氧化石墨烯后,在波數為1 718cm-1處出現的峰歸屬于GO 的C=O 特征峰,在波數分別為1 633 和1 086 cm-1處出現的峰分別是O-H 變形吸收峰和環氧基(C-O)的伸縮振動峰[10],證明氧化石墨烯與SA-PEI成功組合;曲線⑤是改性SA-PEI-GO 的紅外圖譜,比較曲線④和⑤發現,SA-PEI-GO 和改性SA-PEIGO 具有相同位置的特征吸收峰,且這些吸收峰表明凝膠球上含有羥基、羧基和環氧基等含氧官能團。
3.3 改性與未改性對CIP 吸附效果
SA-PEI-GO 凝膠球和改性SA-PEI-GO 凝膠球對CIP 吸附效果對比見圖3。
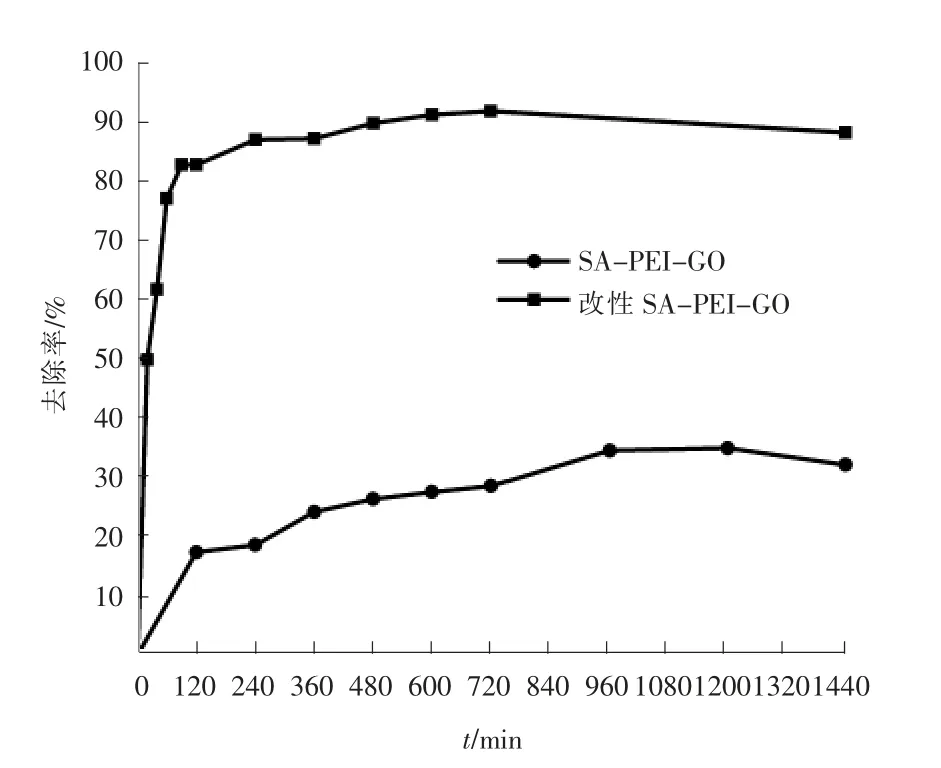
圖3 SA-PEI-GO 凝膠球和改性SA-PEI-GO 凝膠球對CIP 吸附效果
由圖3 可知,改性SA-PEI-GO 和SA-PEI-GO對CIP 去除率分別為91%,34.5%,吸附平衡時間分別為960,600 min,說明碳酸鈣改性可極大地增強SA-PEI-GO 凝膠球對CIP 的吸附性能。
3.4 溶液pH 值對CIP 吸附效果
稱取1 g(濕重)改性SA-PEI-GO 凝膠球分別置于5 組100 mL 質量濃度為2 mg/L 的CIP 溶液中(初始pH 值分別為3,5,7,9,11),考察pH 值對改性SA-PEI-GO 凝膠球吸附CIP 效果的影響,見圖4。

圖4 溶液pH 值對改性SA-PEI-GO 凝膠球吸附CIP 效果的影響
由圖4 可知,改性SA-PEI-GO 對CIP 的吸附效果隨著pH 值增大先高后低,在pH 值為7 時吸附效果最好。 推斷原因是因為當pH 值較低或較高時,吸附劑表面的羥基、 羧基以及胺的官能團發生嚴重的質子化(去質子化)現象,溶液中CIP 呈CIP+(CIP-)形態,吸附劑與吸附質之間強烈的排斥作用降低了吸附效果。 而當pH 值約為7 時,溶液中陽離子形態CIP+和陰離子形態CIP-達到電荷平衡狀態[11],吸附劑與吸附質之間的靜電排斥作用也降至最低,達到最佳吸附效果。 因此,試驗證明最佳pH 值為7。
3.5 材料投加量對CIP 吸附效果
稱取濕重分別為0.25,0.5,1.0,1.5 和1.8 g 的改性SA-PEI-GO 凝膠球分別置于5 組100 mL 質量濃度為2 mg/L CIP 溶液中,在pH 值為7,溫度為25℃的條件下,考察投加量對改性SA-PEI-GO 凝膠球吸附CIP 效果的影響,見圖5。

圖5 投加量對改性SA-PEI-GO 凝膠球吸附CIP 效果的影響
由圖5 可知,改性SA-PEI-GO 對CIP 的吸附效果隨著投加量增大先升高后趨于穩定,投加量為1.8 g(濕重)時吸附效果最好,推斷原因是因為增大投加量可提供更多的吸附活性點位。對此,試驗證明最佳材料投加量為1.0 g(濕重)。
3.6 吸附等溫線和吸附動力學曲線
改性SA-PEI-GO 凝膠球在溫度為25 ℃條件下對CIP 的吸附等溫線見圖6。


圖6 改性SA-PEI-GO 吸附材料對CIP 的吸附等溫線和吸附動力學曲線
由圖6(a)可知,CIP 溶液的初始濃度越高,吸附劑對CIP 的吸附量也越大,原因是由于高濃度促進了液相與固相之間的傳質驅動力,從而增加了CIP分子與吸附劑之間的相互作用[12],但材料的吸附量先快速增加后增速放緩,推斷原因是由于吸附劑表面可利用的活性點位數量減少導致[13]。 由圖6(b)可知,改性SA-PEI-GO 凝膠球在溫度為25 ℃初始質量濃度分別為5,20,50 mg/L 條件下對CIP 的吸附動力學特征。 在前120 min 內,吸附劑對CIP 的吸附速率相對較快,達到最終吸附量的80%以上;隨后吸附速率放緩,在600 min 左右基本達到吸附平衡。其中,第一階段的吸附速率較快是因為材料表面充足的吸附點位及較高的濃度梯度作為驅動力,加速了CIP 分子在材料表面的吸附; 隨著吸附劑表面的活性點位逐漸被CIP 分子占據,固、液界面上的排斥力逐漸增強,導致CIP 的吸附速率降低并最終達到吸附平衡[14]。 因此,試驗證明最佳吸附時間為10 h。Langmuir 模型和Freundlich 模型對實驗結果進行擬合的線性方程式如下:

式中:Ce為CIP 吸附平衡質量濃度,mg/L;qe為吸附劑對CIP 的平衡吸附量,mg/g;qmax為理論最大吸附量,mg/g;KL為Langmuir 常數,L/mg;RL為吸附優惠性指標;KF為Freundlich 常數,L/mg;1/n 為判斷是否為優惠吸附的非均質系數。
Langmuir 和Freundlich 吸附等溫模型的非線性擬合曲線的擬合實驗數據結果見表3。

表3 改性SA-PEI-GO 對CIP 的吸附等溫參數
由表3可知,Langmuir 模型的R2= 0.983,Freundlich 模型的R2= 0.996,對比兩者發現Freundlich 模型的擬合效果更好,說明吸附過程以多層吸附為主;常數RL和1/n 的值均在0~1 之間,說明這2 個模型對吸附過程非常有利[15]。 Langmuir模型中吸附劑對CIP 的最大吸附量為128.11 mg/g,說明該材料對CIP 具有較好的吸附性能。
采用偽一級、 偽二級動力學模型對實驗數據進行擬合的線性方程式分別如下:

式中:t 為某一時刻,min;qe為吸附平衡時單位質量材料對CIP 的吸附量,mg/g;qt為t 時刻單位質量材料對CIP 的吸附量,mg/g;k1為偽一級動力學模型的吸附速率常數,1/min;k2為偽二級動力學模型的吸附速率常數,g/(mg·min)。
偽一級和偽二級動力學模型的非線性擬合曲線的擬合實驗數據結果見表4。

表4 改性SA-PEI-GO 在不同初始CIP 濃度下的吸附動力學參數
由表4 可知,在不同質量濃度(5,20,50 mg/L)CIP 溶液下,偽二級動力學模型(R2=0.991 9,0.996 7和0.997 6)的擬合效果均優于偽一級動力學模型(R2=0.985 2,0.988 3,0.989 4),且偽二級動力學方程計算出的最大吸附量也更接近實驗得到的最大吸附量。 因此,可推斷該吸附過程以化學吸附為主[16]。
3.7 再生性能
改性SA-PEI-GO 吸附CIP 的循環實驗在溫度為25 ℃的恒溫振蕩器中進行,140 r/min 振蕩600 min,采用濃度為0.1 mol/L 的稀鹽酸對吸附劑進行脫附處理。 當投加量為1 g/L(pH 值為7,CIP 質量濃度為2 mg/L,吸附時間為10 h,溫度為25 ℃時改性SA-PEI-GO 對CIP 的吸附可重復利用性見圖7。

圖7 改性SA-PEI-GO 對CIP 的吸附可重復利用性
由圖7 可知,經多次吸附-解吸后,改性SAPEI-GO 凝膠球對CIP 分子仍保持著較好的去除效果,且球體結構并未發生破碎,表明其機械性能良好。
4 結論
(1)將SA,PEI 和GO 混和得到SA-PEI-GO 溶膠體系后加入碳酸鈣粉末,再用氯化鈣交聯形成凝膠球后用鹽酸去除碳酸鈣成功制得改性SA-PEIGO 凝膠球。 改性SA-PEI-GO 和SA-PEI-GO 吸附CIP 去除率分別為91%,34.5%,吸附平衡時間分別為10 和16 h,證明改性SA-PEI-GO 對CIP 吸附性能優于SA-PEI-GO。
(2)通過Freundlich 吸附等溫模型和偽二級動力學模型描述改性SA-PEI-GO 材料對CIP 的吸附過程,說明多層吸附和化學吸附是吸附的主要形式;在吸附劑質量濃度為1 g/L,初始pH 值為7,吸附時間為600 min,吸附溫度為25 ℃時,改性SA-PEI-GO材料對CIP 的理論最大吸附容量為128.11 mg/g。
(3)經過連續6 次吸附-解吸實驗后,改性SAPEI-GO 對CIP 吸附去除率仍達88.4%,且凝膠球無破碎現象,說明改性SA-PEI-GO 凝膠球具有良好的可再生性,是一種具有應用潛力的吸附材料。
猜你喜歡
童話王國·奇妙邏輯推理(2024年5期)2024-06-19 16:03:38
中老年保健(2021年12期)2021-11-30 02:58:01
中學生數理化·七年級數學人教版(2020年10期)2020-11-26 08:24:50
數學物理學報(2020年2期)2020-06-02 11:29:24
攝影之友(影像視覺)(2019年2期)2019-03-05 08:27:14
中華詩詞(2018年11期)2018-03-26 06:41:34
光學精密工程(2016年6期)2016-11-07 09:07:19
Coco薇(2016年8期)2016-10-09 02:11:50
中國塑料(2016年12期)2016-06-15 20:30:07
中國塑料(2015年11期)2015-10-14 01:14:14
