以難治性腹水為主要癥狀的戈謝病1 例
2022-07-12 03:33:54鐘偉志張健成廣東醫科大學附屬第三醫院消化內科廣東佛山528318
廣東醫科大學學報 2022年3期
馬 梅,曹 賢,鐘偉志,張健成(廣東醫科大學附屬第三醫院消化內科,廣東佛山 528318)
提 要:戈謝病是一種罕見的溶酶體貯積病,出現頑固性腹水者則更為罕見。該文報道了1 例因肝脾大、腹水被誤診為自身免疫性肝硬化20 余年自幼行脾切除的戈謝病。
1 病例資料
患者男性,35 歲,因腹脹10 余天于 2020 年11 月30 日入院。患者入院前10 余天無明顯誘因出現腹脹,逐漸加重,以“自身免疫性肝炎后肝硬化”收入院。患者20 余年前在佛山某三甲醫院診斷為“自身免疫性肝炎后肝硬化并脾功能亢進”,于14 歲行脾切除術。近年來多次于佛山市某三甲醫院及我院住院治療,診斷為:(1)自身免疫性肝炎后肝硬化失代償期,大量腹水,低蛋白血癥,脾切除術后;(2)腎病綜合征;(3)高尿酸血癥。體格檢查:體形消瘦,全身皮膚無黃染,未見肝掌及蜘蛛痣,全身淺表淋巴結未觸及腫大,結膜無明顯蒼白,腹膨隆,腹壁靜脈無曲張,腹軟,無明顯壓痛,肝臟捫及不滿意,脾未觸及,移動性濁音(+)。雙下肢輕度水腫。輔助檢查:血白細胞計數 17.65×109/L,中性粒細胞計數 13.01×109/L,鉀 6.46 mmol/L,鈣 1.64 mmol/L,尿素 17.33 mmol/L,肌酐 144 μmol/L,白蛋白 13.7 g/L,谷草轉氨酶 57 U/L,堿性磷酸酶 423 U/L,膽堿脂酶 3 678 U/L,尿潛血+++,尿蛋白定性+++,尿微量白蛋白 >150 mg/L,24 h 尿蛋白定量5 756 mg/24 h。入院后予補充白蛋白、利尿、改善肝功能等治療后,腹水消退不明顯,且腹水反復迅速增加,低蛋白血癥糾正欠佳。行骨穿檢查未能抽出骨髓液,骨髓干抽考慮骨髓纖維化。骨髓涂片:增生尚活躍骨髓象,粒系比例增高,血小板呈大簇狀分布。骨髓活檢:骨髓纖維化(MF-3級、膠原纖維-2 級、骨硬化-0 級)。HLA-B27-DNA 陽性(+)。JAK2 基因突變陰性。紅細胞CD55、CD59 陰性。抗磷脂酶A2 受體抗體陰性。腹水細胞學檢查未見癌細胞。雙側腎靜脈彩超未見異常。于2021 年1 月4日再次入我院腎內科住院治療,住院期間在B 超引導下行右腎穿刺活檢術。腎臟病理診斷:(1)膜增生性腎小球腎炎;(2)腎組織內散在皺紋紙樣組織細胞見圖1、2。考慮高雪細胞(戈謝病)可能性大,行戈謝病GBA基因測序(圖3):檢測到基因變異。β 葡萄糖苷脂酶活性為1.95 nmol/(h·mg),遠低于參考指標6.8 nmol/(h·mg)。腎穿及戈謝病GBA 基因檢測均提示戈謝病可能,β 葡萄糖苷脂酶活性降低大于30%,戈謝病診斷明確。確診I 型戈謝病后建議應用酶替代治療(enzyme replacement therapy,ERT),由于患者經濟無法承擔未行ERT,給予口服嗎替麥考酚酯膠囊0.5 g,每天2 次,聯合激素潑尼松50 mg,每天1 次,以減少蛋白漏出,輸注人血白蛋白及食用高蛋白食物提高蛋白質攝入,呋塞米利尿。4 個月后患者白蛋白提升明顯,復查白蛋白29.6 g/L,腹水消退。患者腹水的顏色比普通肝硬化患者腹水顏色澄清,達到幾乎與生理鹽水一致的顏色。
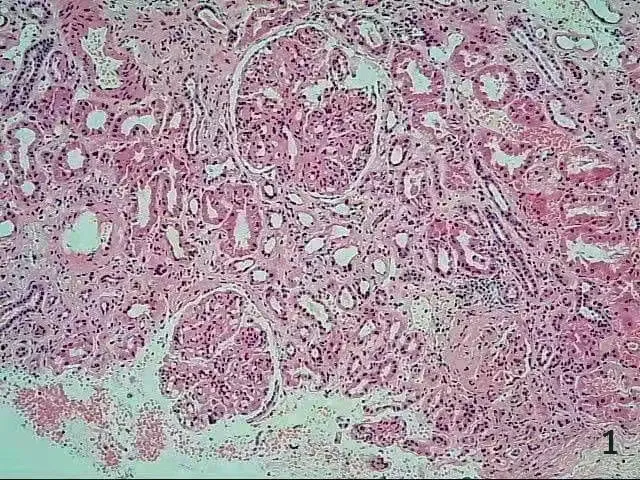
圖1 戈謝細胞(HE 染色×100)
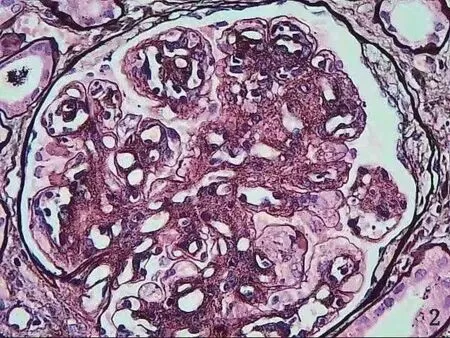
圖2 戈謝細胞(PASM 六胺銀染色×400)
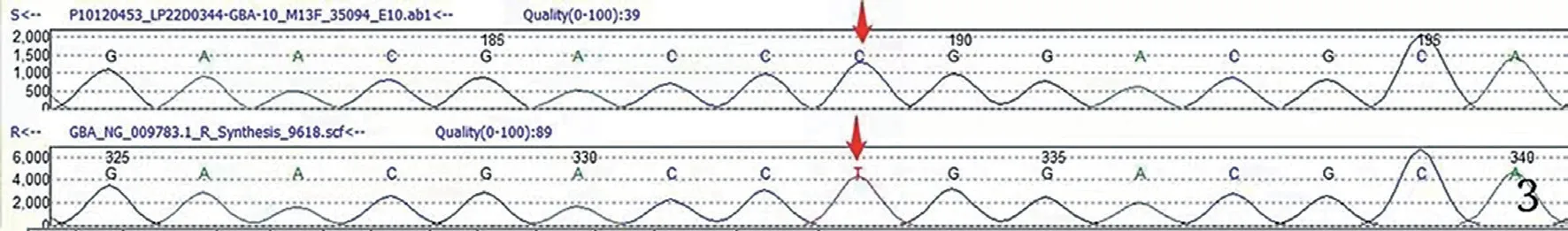
圖3 基因測序檢測到基因變異峰圖
2 討論
戈謝病又稱高雪病、高歇病、葡萄糖腦苷脂沉積病,是一種罕見的溶酶體貯積病,為常染色體隱性遺傳代謝病[1]。該病主要發病機制是體內編碼β-葡萄糖腦苷脂酶的基因突變,致患者巨噬細胞中溶酶體內該酶缺乏或降低[2],造成其底物葡萄糖腦苷脂在肝、脾、腎、骨骼、肺、甚至腦的巨噬細胞中貯積,形成典型的貯積細胞即戈謝細胞[3],從而引起以顯著肝脾腫大、貧血、骨質疏松及神經系統受損等臨床表現為特征的疾病。臨床上分為3 種類型,I 型(非神經病變型):約占戈謝病的90%~95%,臨床上主要表現為肝脾腫大,常伴有脾功能亢進,血液學表現為血小板減少和貧血。而此型患者中累及腎臟導致低蛋白血癥、頑固性腹水者罕見。Ⅱ型(急性神經病變型):臨床上主要表現神經系統受累,嬰幼兒期發病,一般2~3 歲前死亡。Ⅲ型(亞急性神經病變型):常發病于兒童期,早期表現與I型相似,逐漸出現神經系統受累表現,伴發育遲緩、智力落后,病情進展緩慢[4]。戈謝病臨床上常用的實驗室檢測有葡萄糖腦苷脂酶活性檢測、骨髓細胞學監測、殼三糖酶活性監測和基因檢測,其中葡萄糖腦苷脂酶活性檢測是戈謝病診斷最有效、最可靠的方法,當外周血白細胞或皮膚成纖維細胞中葡萄糖腦甘酯酶降低至正常值的30%以下時,即可確診戈謝病[5]。戈謝病的治療主要包括酶ERT、造血干細胞移植、底物抑制療法。ERT 作為戈謝病的一線治療藥物已有20 余年,可糾正貧血、血小板減少,使肝脾體積回縮,體格發育,骨痛緩解,從而提高其生存質量,但需經靜脈給藥,并且費用較高,需終生使用[6-7]。目前各種指南及共識僅推薦ERT 用于I、Ⅲ型戈謝病患者。若Ⅱ型戈謝病患者ERT效果差,可行脾切除、分子伴侶療法、骨病的支持治療、基因療法及對癥治療的非特異性治療。
戈謝病臨床罕見,本例幼時起病,以肝脾大為主要表現,誤診為肝硬化多年,成年后因合并嚴重腹水反復就診效果欠佳,行腎穿發現戈謝細胞診斷戈謝病。本例患者成年后嚴重腹水考慮為葡萄糖腦苷脂在腎臟貯積導致腎小球濾過膜受損,形成大量蛋白尿所致。此案例提醒我們凡是診斷腎病綜合征的患者一定要行腎穿刺,以明確病理分型,和排除戈謝病等繼發性因素。臨床上能夠引起腹水的病因有很多[8],而同時伴有肝脾大的常見于肝硬化,但肝硬化失代償期肝臟基本縮小。此病例肝臟持續性增大,與大部分肝硬化失代償期患者癥狀不符合,且患者無門體側支循環的形成及門脈高壓性胃病,與典型肝硬化失代償期表現不吻合。臨床上遇有大量腹水、脾大、低蛋白血癥與肝硬化失代償期癥狀相類似的患者,除了考慮肝硬化同時需考慮戈謝病可能,應進一步行肝穿刺活檢以明確診斷。
