基于化學發光法的高純氣體中ppb量級NOx 濃度測量*
2022-07-22 05:57:02張猛彭志敏楊乾鎖丁艷軍杜艷君
物理學報 2022年13期
張猛 彭志敏 楊乾鎖 丁艷軍 杜艷君
1) (華北電力大學控制與計算機工程學院,北京 102206)
2) (中國科學院大學工程科學學院,北京 100049)
3) (中國科學院力學研究所,流固耦合系統力學重點實驗室,北京 100190)
4) (清華大學能源與動力工程系,電力系統與發電設備控制與仿真國家重點實驗室,北京 100084)
1 引言
隨著科技的發展,社會生產對高純氣體的需求日益增加,尤其是在航空航天、半導體、等離子體干刻、大規模集成電路及燃料電池等行業[1,2].然而,受限于氣體的提純方式,高純氣體中仍存在多種雜質成分,主要包括水、氧氣、硫化物、氮化物等痕量氣體[3].氣體雜質成分對加工精度具有顯著影響.如,在質子交換膜燃料電池工作時,高純氫氣攜帶的痕量雜質氣體(H2S,SO2)對陰極催化劑具有毒化作用,產生不可逆的破壞行為[4];雜質痕量氣體也是影響大規模集成電路生產質量的關鍵因素,不僅會直接影響晶體的生成速率,而且一定程度上決定了結構的應變分布[5],導致結構缺陷.因此,對高純氣體中關鍵痕量雜質氣體的濃度進行檢測尤為必要.
不同雜質氣體的定量檢測手段因其特性而存在差異,其中熱導池(TCD)、氦離子化(HID)、脈沖放電氦離子化(PDHID)、質譜法(MS)方法具有較強普適性,可以實現絕大多數永久性氣體組分濃度的檢測.對于常見影響加工精度的雜質氣體,測量方法更為多樣,如O2雜質組分,通常借助電子捕獲(ECD)、微量O2分析法、調制二極管激光吸收光譜法(TDLAS)、質譜法等方法進行定量測量[6],其中TDLAS 法借助Herriott 池等裝置可實現ppb(1 ppb=10—9)量級的檢測,測量精度相對較高[7,8].對于硫化物雜質的定量檢測,常采用火焰光度法(FPD),基于原子光譜理論和光電轉換測量裝置實現ppm(1 ppm=10—6)量級的檢測,但高純氣體中的硫化物含量一般在ppb 量級,利用該檢測手段很難進行標定.相比于硫化物檢測,ppb 量級含氮化合物的檢測可以借助熱離子化(TID)實現,檢測限為10—13—10—8g/s,但其只能檢測物質的含氮總量,難以對不同氮化物分子(NO,NO2,HNO3···)進行選擇性識別,因此對于NOx(NOx=NO +NO2)氣體檢測并不適用[9];TDLAS 法雖對NOx氣體具有高選擇性,但是難以實現亞ppb 量級測量,所以探究高純氣體中ppb 量級的硫化物、氮化物等微量雜質氣體高識別性、高靈敏度的檢測方法具有重要意義.
相比于上述檢測手段,化學發光法具有高靈敏度、低檢測下限、易操作性等優點[10-12],可以實現高純氣體中ppb 量級的氮化物(硫化物)雜質組分的快速檢測.該測量方法遵循發射光譜原理,當NO(SO 中間體)與O3發生有效碰撞時發生氧化反應,通過化學激發方式生成激發態的NO2(SO2)分子,該分子不穩定,迅速退激發至基態并對外輻射光子[13],對應的輻射光子數與參加反應的粒子數成正比,通過弱光探測器探測相應的積分光強信息進而反演粒子數濃度,最終實現痕量雜質氣體的低濃度檢測[14].1966 年,Clough 等[15]發現激發態NO2退激發時會對外輻射光子并用單色儀確定相應波長范圍(600—3000 nm,中心波長約為1200 nm),為分子光譜的識別及診斷提供了理論依據.1970年,Fontijn 等[16]通過優化測量裝置獲得了4 ppb NO 的化學發光信號,初步實現了低濃度NO 的測量.1989 年,Ridley 等[17]通過大功率真空泵降低反應腔真空度以減弱淬滅效應影響,實現了1 ppt的NO 測量下限,使得該測量方法的檢測下限得到進一步突破,但是大流量真空泵由于重量大、壽命短、操作復雜等原因,并未得到廣泛應用.為了進一步實現其他形式的氮化物(NO2,HNO3···)的測量,20 世紀末,研究學者對相應的轉換機制進行了大量研究[18].Kliner 等[19-21]發現在高溫環境中,NO2可以被金屬(Au,Ag,Pt,Mo)催化還原成NO 產物,同時,Bollinger 等[22]探究了HNO3及其他氮氧化物在高溫金管內被CO 氣體催化轉換成NO 的規律,實現了其他形式含氮化合物的間接測量.
本文首先結合高比表面積、高孔隙率鉬網設計形成NO2催化轉化反應器(轉化率η> 98%),自主設計搭建高精度、低檢測下限的ppb 量級濃度的NOx實時測量系統;然后,利用氮氣稀釋的標氣對自主設計的弱光探測系統進行標定,并綜合考慮不同背景氣體的熒光、磷光淬滅效應,建立常見氣體背景條件下NOx濃度測量方法;最后,利用所建立的測量理論及系統對4 種常見高純氣體中痕量NOx氣體進行測量,并結合氣體的制備及提純工藝對高純氣體中的ppb 量級NOx濃度含量進行評價,旨在為燃料電池、半導體器件制備等尖端科技領域提供可靠的雜質氣體成分診斷信息,降低痕量雜質氣體對加工工藝的影響.
2 理論基礎
2.1 化學發光理論
在常溫低壓條件下,NO 分子與O3分子發生有效碰撞時生成激發態的[23],該激發態產物穩定性差,會迅速退激發至基態并對外輻射特定波段的光子.當臭氧濃度過量時,輻射積分光強與NO 濃度呈現線性相關規律[24],故可以利用弱光探測器(PMT)對其化學發光光強進行探測,進而獲得NO 濃度.
化學發光詳細反應機理如圖1 所示.化學激發產生單線態形式(Si)的激發態電子,基于Frank-Condon 原理[25],激發能級上的電子通過振動弛豫方式轉移至勢能面極小值處,然后進行輻射躍遷回到基態并對外釋放光子(熒光)[26];在自旋耦合作用下,電子在單線態能級和三線態能級交叉點處,其自旋方向改變發生系間竄越,從單線態能級轉移到三線態能級,然后通過振動弛豫到達能級勢能面的極小值位置,最后以光子輻射形式退至基態(磷光).以上兩種退激發輻射光子波長范圍覆蓋可見光、近紅外及中紅外波段(600—3000 nm),但由于PMT光譜響應波段多為可見光及近紅外范圍(600—1200 nm),可利用兩者的交叉光譜區域進行NOx測量.此外,退激發時存在無輻射現象,激發態分子或直接與第三體(O2,N2,O3···)碰撞,發生無輻射形式的能量轉移淬滅[27].當單線態能級與基態能級能量相同時,激發態電子會以振動弛豫方式進入基態能級并在能量極小值位置以聲子及熱的形式到達基態,該過程無光子輻射.以上物理過程涉及的主要化學反應方程式如(1)—(4)式所示[28]:
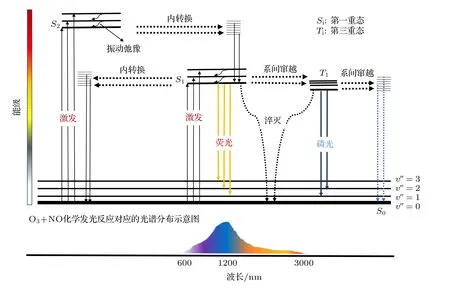
圖1 NO 與O3 化學發光反應能級躍遷原理示意圖Fig.1.Schematic diagram of chemiluminescence reaction level transition of NO and O3.
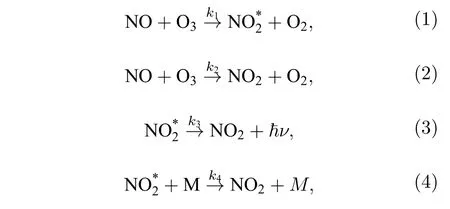
式中,ki是化學反應速率常數,用于表征化學反應速率快慢,由實驗測得[26,29].M是參與碰撞淬滅的第三體(N2,O2,O3,CO2,Ar···).(1)式是NO 與O3反應的主要途徑,其效率由反應室體積、結構、壓強及氣體流速、溫度和臭氧濃度等物參決定[30].(2)式為少量分子的無輻射退激發行為.(3)式中激發態NO2退激發輻射光子行為受到(4)式影響[29],其中(4)式為激發態分子退級時的淬滅行為.當反應腔壓力減小時,粒子數體密度降低,NO2*與第三體碰撞概率降低,淬滅現象減弱.但氣流流速過快時,NO 分子在反應室滯留時間縮短,可能會降低(1)式的反應效率,綜合各種因素,化學發光的探測光強如(5)式所示[16]:
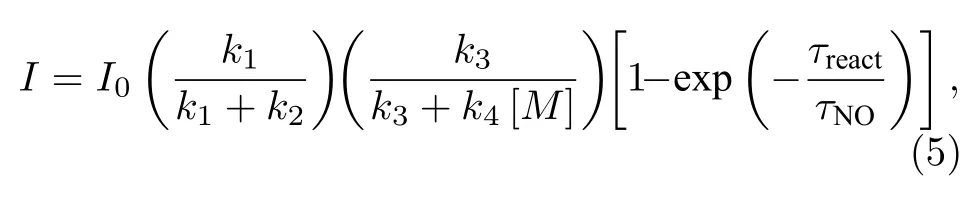
式中I是弱光探測器檢測到的實際光強,I0是NO分子僅通過(1)式、(3)式反應途徑時的理論輻射光強.τreact是NO分子在反應腔的殘余壽命,τNO是NO 分子的反應壽命.當探測系統的結構和實驗參數一定時,探測光強I值與稀釋平衡氣體的種類有關,故本文在實際NO 濃度測量時考慮了不同平衡氣體引入的淬滅效應[27].
3 實驗裝置
3.1 測量系統
化學發光NOx探測系統裝置如圖2 所示,主要包括NO2催化轉化模塊、等離子體(空氣源)、長方體反應腔、弱光探測模塊及Labview 信號采集模塊.NO2催化轉化模塊是由高比表面積、高孔隙率鉬網多層堆積而成,可以在350 ℃高溫條件下,將NO2催化還原被還原成NO,還原效率高于98%.在高溫條件下,高價氮氧化物、硝酸等其他雜質組分也會參與催化還原反應生成NO,但考慮以上氣體在高純氣體中含量相對NOx濃度較低,因此本文認為Mo 催化轉化后NO 來自于NO2雜質組分.在實際測量過程中,等離子體為化學發光反應提供過量臭氧,本文臭氧制備采用高壓介質阻擋放電方式,在玻璃材質的圓筒內外粘貼鋁箔電極,放電間距為1.25 mm,介質厚度為1.5 mm,當電極間脈沖電壓為6 kV,頻率為80 Hz、占空比為1.7% 時,流速為70 mL/min 的空氣流經板間縫隙時發生電離(圖中藍色區域),產生約1000 ppm 的臭氧,后經流量控制模塊進入反應腔(如藍色箭頭所示).由于樣氣路和反應腔內的氣體殘留導致系統響應產生滯后,故反應腔不宜過大,本文采用小型長方體形狀腔體,尺寸為8 m m×8 m m×20 mm,為了提高反應腔的化學反應效率,兩路氣體在入口處采用套管混合方式,并且將出氣口遠離進氣口以提高氣體殘余壽命.同時為了提高光收集效率,內壁貼有鍍銀高反射平面鏡.化學發光光強通過弱光探測系統進行探測,為了避免雜散光的干擾,在PMT 探測靶面前放置紅光濾波片(恒洋光學,HGLP),同時為了降低PMT 本身熱噪聲干擾,利用珀爾帖對其進行制冷,溫度控制在 —6 ℃左右.弱光探測信號輸出時,通過跨阻放大方式將光電流信號轉換成電壓信號,再結合信號放大電路來增加信號的信噪比以提高系統的探測下限,然后借助 Labview 軟件和采集卡(ART Technology,8502)硬件等對信號進行實時采集和處理.
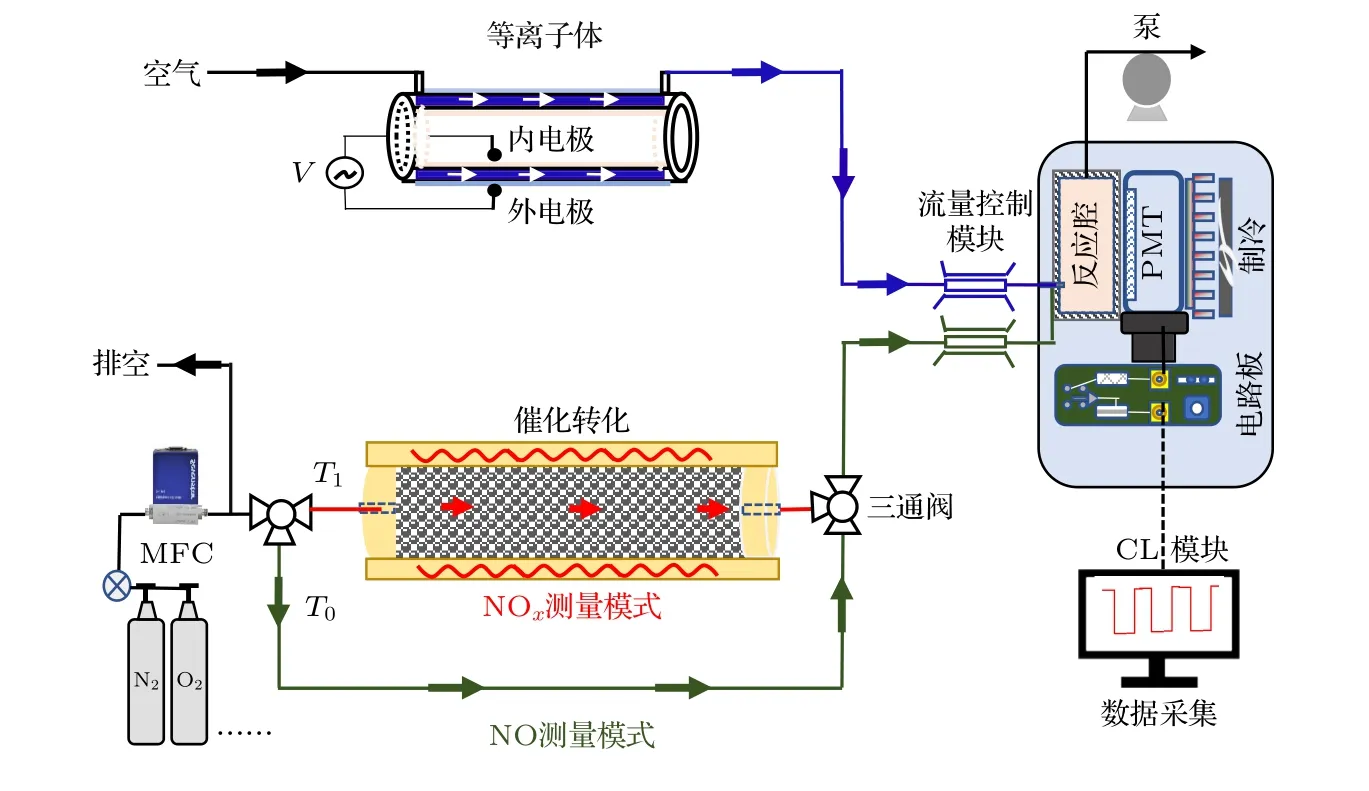
圖2 化學發光測量系統裝置示意圖.V 為可調節脈沖型高壓電源;MFC 為流量控制計;CL 為化學發光;PMT 為光電倍增管;T0 為NO 測量模式通道;T1 為NO2 測量模式通道Fig.2.Schematic diagram of chemiluminescence measurement system.V is adjustable pulse high voltage power supply;MFC is flow control meter;CL is chemiluminescence;PMT is photomultiplier tube,T0 is NO measurement mode channel;T1 is NO2 measurement mode channel.
3.2 測量邏輯及方案
為了實現NO/NOx同步測量,首先需確定測量系統的背景信號.切斷等離子體的高壓電源,打開真空泵保持反應腔穩定在負壓狀態,樣氣路和臭氧路的氣體通過流量控制模塊穩流后進入反應腔,進氣流量分別為70 mL/min 和700 mL/min,此時弱光探測系統的電壓信號記作測量系統的背景信號.該基線由系統的機械振動、氣體擾動、反應腔內雜散光、PMT 熱噪聲、電路板內電流噪聲等多種因素決定,計算NOx濃度值時需將其扣除.獲得穩定的背景信號后,依次進行NO/NOx測量.打開等離子的高壓電源,電離產生的臭氧經過聚四氟乙烯管,迅速進入反應腔與NO 氣體發生化學反應退激發光,其光信號經過弱光探測系統轉化成電壓信號.當樣氣路的三通閥切換至T0時(綠色氣路),進入NO 測量模式,該模式持續10 s,由數字式計時器自動控制,對應NO 信號如黑色短劃線部分所示,將其扣除背景信號后即為NO 信號.當三通閥自動切換為T1時(紅色氣路),待測樣氣進入高溫加熱的鉬催化轉化器中,NO2在高溫條件下會被Mo 催化還原成NO,經過還原的樣氣流經聚四氟乙烯管路進入反應腔與臭氧混合,對應的化學發光信號通過弱光探測系統轉換成電壓信號,該信號扣除NO 信號后可得NO2信號.以上NO/NOx測量模式通過三通閥進行動態切換,不同模式下的化學反應弱光信號借助采集卡硬件及Labview 軟件等實現動態采集,采樣間隔設置為1 s,故可以得到方波分布形式的電壓信號.
如圖3 為化學發光探測系統對63.3 ppb NO2和39 ppb NO 混合標氣的動態測量結果.關閉臭氧時的背景信號約為0 mV,當測量系統切換至NO測量模式時,混合樣氣中的NO 電壓信號約為3 mV,明顯高于背景信號幅值,說明該測量系統的靈敏度高,測量下限低.在NO2測量模式下,NOx混合氣體經過鉬催化轉化器后NO 電壓信號約為9 mV,扣除催化轉化前NO 電壓信號即得NO2電壓信號,約為6 mV,信噪比約為40.在模式切換時,測量系統響應迅速,約為1 s,對于可以用于ppb 量級NOx濃度的實時檢測.
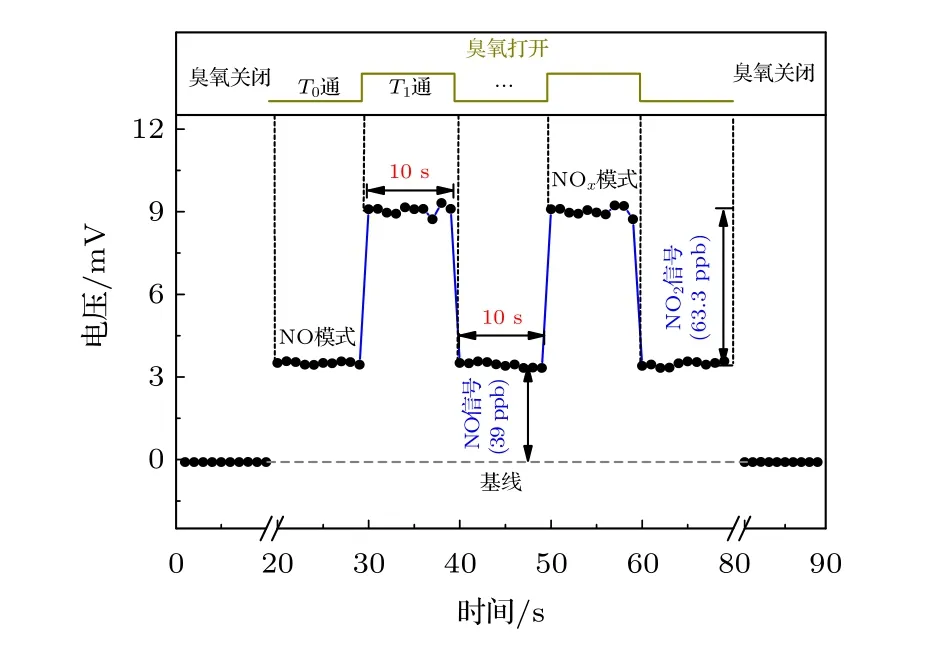
圖3 化學發光探測系統對63.3 ppb NO2 和39 ppb NO 混合標氣的動態測量結果Fig.3.Dynamic measurement results of 63.3 ppb NO2 and 39 ppb NO mixed standard gas by chemiluminescence detection system.
4 結果與討論
4.1 測量系統性能
本文對NOx測量系統的綜合測量性能進行探究,通過標定實驗和Allan 方差分析來獲取測量系統的線性響應系數及探測下限.基于稀釋法,用高純N2對NO2標氣(10 ppm,氮氣平衡,)進行稀釋,以制備不同濃度的NO2標氣,濃度范圍為63.3—605 ppb,共10 個濃度點.測量方式如 3.2 小節所述,每種工況測量時間為10 s,每個濃度工況重復測量5 次,取其平均值作為濃度測量值.圖4(a)為測量原始信號,在濃度一定時,電壓信號由于三通閥周期性切換而呈現方波分布規律,背景噪聲信號幅值約為 —0.9 mV,雖然測量系統在標氣濃度增大時,可能會引入更多的光電流噪聲影響,但這對背景信號貢獻很小,故在假設系統溫漂很小及其他慢變噪聲成分很低時,近似認為初始背景信號值不隨時間而變化.圖4(b)表明,不同NO2濃度下的化學發光信號與其濃度呈現強相關性,其擬合相關系數R2為0.99967,線性關系式為y=0.093x-0.458(y為電壓信號,x為NO2濃度).在氣體擾動及PMT光電流等因素干擾下,NOx的電壓信號會產生微小波動,當NO2濃度為370 ppb 時,信噪比約為383.測量系統線性度受到鉬催化轉化器轉換效率、NO 反應效率、光收集效率等諸多因素影響,當鉬催化轉化器轉化效率不穩定時,NO2的轉化系數不再是常數,由于線性系統不穩定性傳遞關系,濃度輸入量和電壓輸出量不再呈現線性相關規律,結合高線性度的標定結果可推知本文鉬催化轉化器的轉化效率較穩定.此外,當臭氧量不足時,NO 分子在反應室駐留階段與O3分子的有效碰撞概率降低,導致探測系統NO 捕捉能力減弱,探測光強隨之呈現非線性分布,線性結果再次說明本文選取的臭氧濃度(1000 ppm)對于測量系統的線性度而言比較合理.
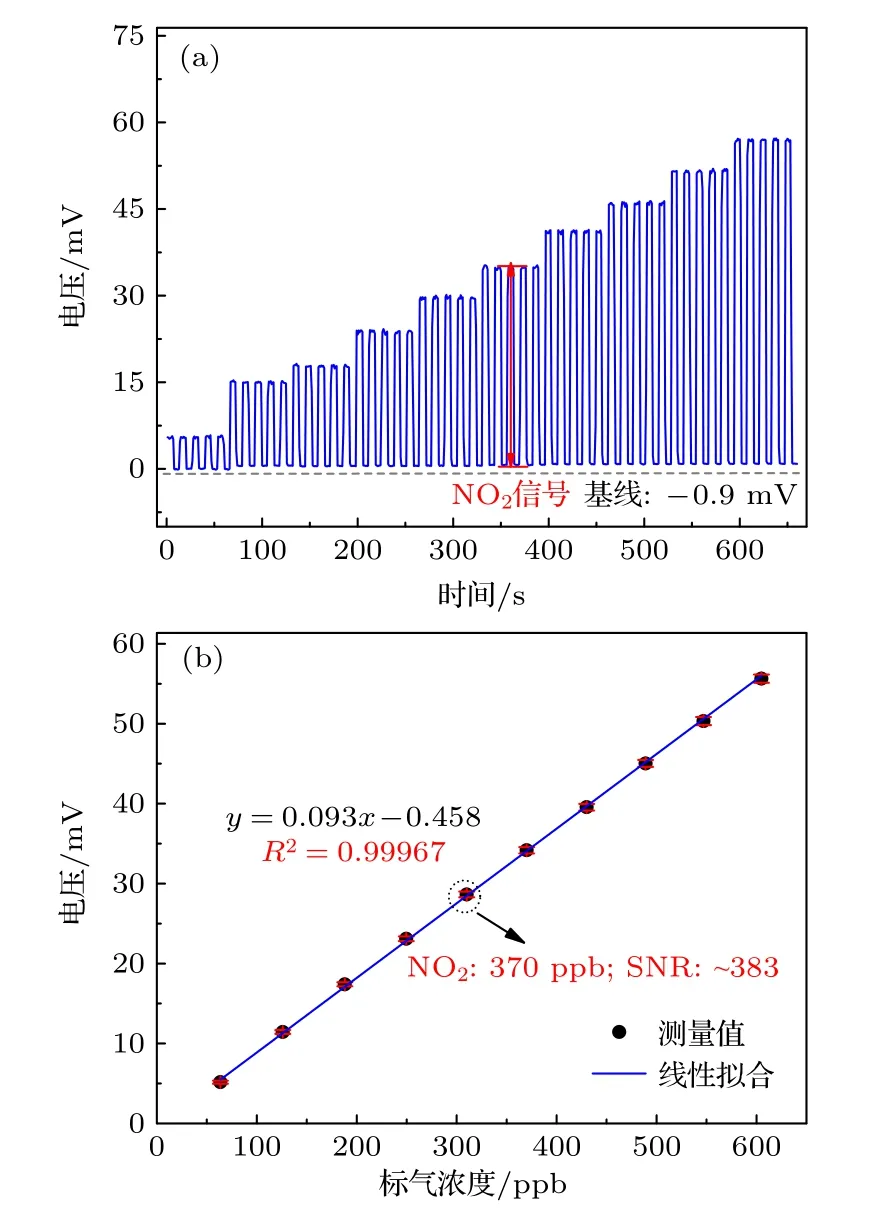
圖4 不同NO2 標氣濃度測量結果 (a)不同NO2 濃度下對應5 個周期的原始測量信號;(b)不同NO2 標氣濃度的電壓信號及其線性擬合曲線Fig.4.Different NO2 standard gas concentration measurement:(a) Original measurement signals corresponding to 5 cycles at different NO2 concentrations;(b) voltage signals of different NO2 gas concentrations and their linear fitting curves.
為了確定系統的測量下限,關閉數字式計時器,切換至NOx持續測量模式,依次對空氣及“零氣”(經過活性炭和高錳酸鉀過濾的空氣)進行30 min 測量,結果如圖5 所示,在積分時間為1 s 時,相比于“零氣”源,空氣源的Allan 方差均方根更高,噪聲水平增加的主要原因為空氣中的NO2濃度較高(10—20 ppb),化學發光信號增強時會使PMT 的光電流噪聲增大,該噪聲符合泊松分布規律,由于其頻率波段較高,可以通過延長積分時間方式進行消除.當積分時間延長至15 s 時,光電流噪聲影響基本被消除,空氣和“零氣”源的噪聲水平相當.隨著積分時間的延長,白噪聲等其他高頻噪聲影響進一步被消除,當積分時間為285 s 時,探測系統獲得最低檢測下限,檢測值為25 ppt (1σ),當積分時間繼續延長時,系統中溫度漂移等慢變噪聲成為噪聲主導因素,基線漂移會引入測量誤差,導致系統測量精度有所降低.
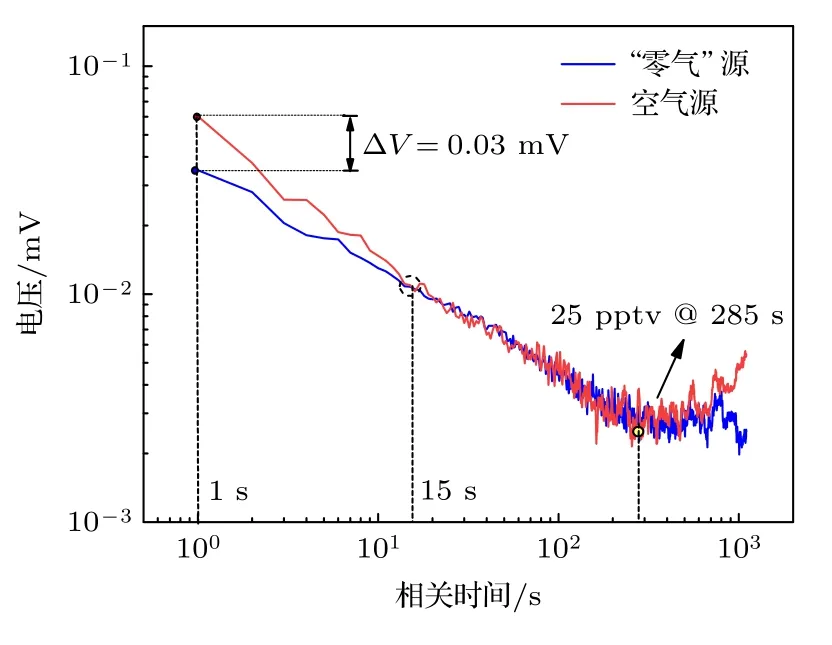
圖5 “零氣”和空氣源時測量系統電壓信號的Allan 標準差Fig.5.Allan standard deviation of the voltage signal of zero gas and air for the measurement system.
4.2 高純氣體中NOx 濃度淬滅效應修正
在NOx測量系統中,反應腔內NO 的化學發光光強不僅與NO 粒子數有關還與平衡氣體的種類有關,當NO 濃度恒定時,平衡氣體種類是影響化學發光效率的重要因素.在反應腔內,化學反應生成的分子退激發時會對外輻射光子(熒光、磷光)或與第三體碰撞發生淬滅.其中熒光壽命相對較短,約為10—9—10—6s,在激發態電子躍遷回基態過程中,分子與第三體發生碰撞淬滅或能量轉移淬滅概率較低,能量消耗較小.但對于磷光而言,系間竄越行為需要的周期較長,約為10—4—10 s,磷光淬滅現象顯著增加,成為影響化學發光效率的主要因素.碰撞淬滅或能量轉移淬滅過程與平衡氣體的黏性及其分子能級分布相關,即當NOx濃度一定時,背景氣體種類發生變化,化學發光效率亦會隨之改變.因此,在利用化學發光法對不同背景氣稀釋環境下的ppb 量級NOx濃度進行檢測時,需要考慮背景氣體種類改變導致的化學發光淬滅效應差異,避免引入測量誤差.
對于特定的第三體氣體,相應的碰撞淬滅和能量轉移淬滅效應相互耦合,難以對其各物理過程中的淬滅系數進行獨立測量,故在本文中,只考慮由于氣體組分不同而引入的綜合淬滅系數差異.在保證稀釋氣體中NOx摩爾體積比一定時,依次改變第三體氣體的摩爾體積比以探究不同第三體氣體相對N2的淬滅系數,結果如圖6 所示.相比于氮氣,氬氣的淬滅現象減弱,當氬氣摩爾體積比例為100%時,實驗測得的相對淬滅系數約為0.73,即化學發光信號相對增大約27%.相比而言,CO2的淬滅效應明顯,當平衡氣體完全替換成CO2時,相對淬滅系數增加至2,相應光強減弱至原來的50%.此外,氧氣的相對碰撞系數僅有約1.115,與氮氣接近.故當背景氣體種類發生改變時,需要考慮不同氣體化學發光淬滅效應的差異,對測量結果加以修正.
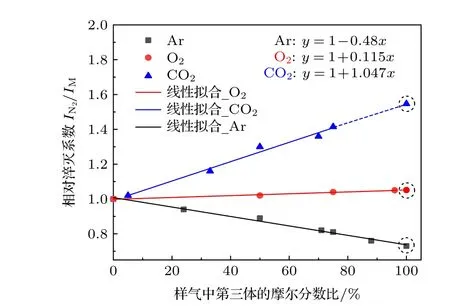
圖6 第三氣體的相對淬滅系數[31]Fig.6.Relative quenching coefficient of the third gas[31] .
4.3 高純氣體中NOx 濃度測量
硅烷、鍺烷、磷烷、砷烷、三氟化硼作為半導體及燃料電池行業常用氣體,其中鍺烷、磷烷、砷烷、三氟化硼高純氣體中NOx雖然理論上可以借助化學發光探測系統實現測量,但考慮到催化還原裝置壽命及人員安全問題,本文僅基于上述測量系統及建立的高純氣體測量理論和方法,對常用的高純氣體(N2:99.999%;Ar:99.999%;O2:99.5%;CO2:99%)中NOx濃度進行定量測量.
實驗時,借助流量控制模塊保證高純氣體的進氣量相同,此時,化學發光光強由高純氣體中NOx雜質濃度及淬滅效應決定.測量結果主要如圖7 所示,黑色虛線之間的信號差值源于樣氣中NO2成分(紅色箭頭)或NO 成分(藍色箭頭),如,純O2中NO2電壓信號為0.3 mV,NO 電壓信號為0.11 mV,信噪比分別為3 和1.其中空氣中NO2含量最高,主要是由于車輛尾氣、工業排放所致[32].“零氣”是將空氣經過活性炭和高錳酸鉀進行過濾處理后的氣體,其中的氮氧化物組分會被有效脫除,因此相比于空氣測量結果,其NOx含量很低,NO 及NO2濃度僅有2 ppb 左右,如表1 所示.
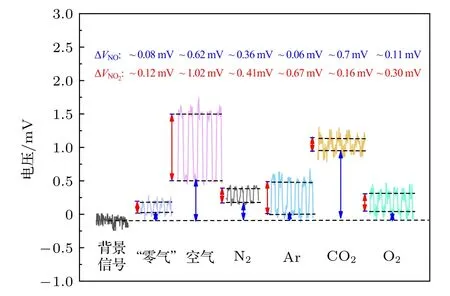
圖7 空氣、“零氣”及高純氣體中NOx 濃度測量Fig.7.Measurement of NOx concentration in air,zero gas and high purity gas.
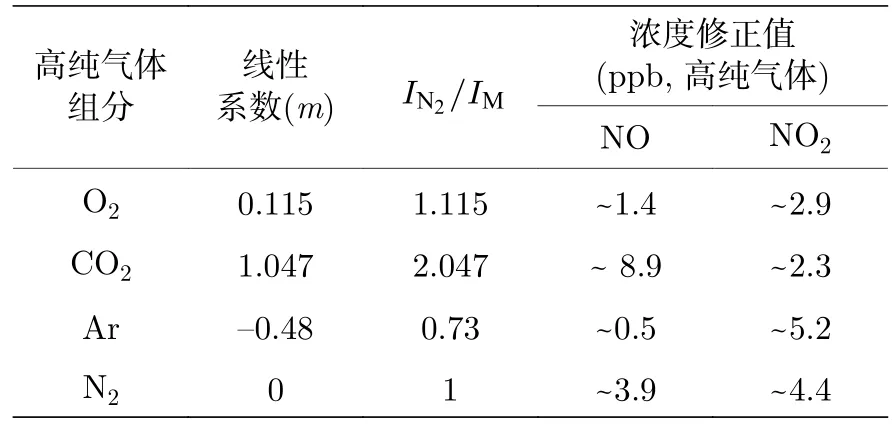
表1 不同組分高純氣體的NOx 轉換系數及其測量結果修正值Table 1.NOx conversion coefficient of different high-purity gas components and modification of measurement results.
基于不同平衡氣體的測量方法,高純氣體中NOx測量結果如表1 所示.CO2中NO 的含量相對較高,約為9 ppb,相比于Ar,O2,N2的空氣液化方式,其提純流程更為復雜:首先需在煙氣、化工尾氣等惡劣環境中利用堿液進行碳捕捉,該環節會吸收較多NOx雜質氣體,隨后在高壓低溫環境中(7—8 MPa,—20 ℃左右)進行液化處理時,NOx雜質亦會隨之發生物態變化,在進一步的蒸餾解析過程中(溫度升高,壓力降低),CO2揮發會攜帶混合液體的痕量NOx氣體,由于NOx的沸點和CO2沸點不同,CO2高純氣體中的NOx絕對含量很低(ppb 量級),但導致高純CO2氣體中的NOx含量相對偏高.其他3 種高純氣體均是通過直接空氣液化再精餾得到,因為空氣中的NOx濃度含量很低,故提純后的NOx雜質氣體濃度偏低.在純氧環境中,由于其氧化作用,雜質NO 殘留較少,僅為1.4 ppb左右,相應NO2濃度會有所增大,約為2.9 ppb.在純Ar 氣體中,NO 含量很低,約為0.5 ppb,但其 NO2濃度含量偏高,約為5.2 ppb,這可能與氣體精餾時的溫度和壓強有關.在純氮體系中,NO含量約為3.9 ppb,NO2含量約為4.4 ppb,故用純氮氣對NO 標氣進行高倍稀釋時,需要考慮平衡氣體引入的濃度稀釋誤差.如在將濃度為10 ppm NO 利用高純氮氣稀釋至20 ppb 時,高純氮氣中的2 ppb NO 雜質會引入約10%的稀釋誤差.本文在利用稀釋法對標氣進行稀釋時,考慮了高純N2系統中ppb 量級的NO 雜質氣體引入的稀釋誤差并加以修正,進而提高系統測量的準確性.
5 結論
本文基于化學發光光譜理論和催化轉化機理設計了一套氮氧化物測量系統,并通過氣路自動切換方式實現ppb 量級NOx濃度的實時測量.標定實驗結果表明,系統對氮氣平衡的不同濃度NO 標氣線性響應度高,其擬合相關系數R2約為0.99967,同時對系統進行空氣和“零氣”源的Allan 方差分析表明,當積分時間約為285 s 時,系統的慢變和快變噪聲綜合影響最小,最低探測下限約為25 ppt,結果說明化學發光法相比于其他氮氧化物(硫化物)測量方法,具有高信噪比、低探測下限等優勢.隨后綜合考慮不同背景氣體的淬滅效應,建立不同背景氣體的測量理論和方法,借助上述系統對常見的4 種高純氣體中的ppb 量級的氮氧化物進行測量,其中CO2中的NO 含量較高,約為9 ppb 左右,其他高純氣體中的NO 含量均很低(< 4 ppb),主要由于CO2提純不同于其他3 種高純氣體的空氣液化方式,該氣體需要在煙氣、化工尾氣中進行初步碳捕捉后再精餾.綜上,化學發光法可以實現高純氣體中ppb 量級的NOx濃度測量,為降低高純氣體中雜質成分對加工工藝影響提供可靠、必要的濃度診斷信息.
猜你喜歡
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
鴨綠江(2021年35期)2021-04-19 12:24:18
考試與評價·高一版(2020年6期)2020-11-02 02:45:24
中學生數理化·八年級物理人教版(2019年9期)2019-11-25 07:33:02
中學生數理化·八年級物理人教版(2019年3期)2019-04-25 06:20:54
電子制作(2018年11期)2018-08-04 03:25:42
中學生數理化·八年級物理人教版(2018年3期)2018-05-31 08:52:45
海峽科技與產業(2016年3期)2016-05-17 04:32:12
