十四烷微膠囊的制備及性能研究*
2022-08-03 11:23:44常洋琿孫志高
功能材料 2022年7期
關(guān)鍵詞:體系
常洋琿,孫志高
(蘇州科技大學(xué) 環(huán)境科學(xué)與工程學(xué)院,江蘇 蘇州 215009)
0 引 言
經(jīng)濟(jì)快速發(fā)展的同時不可避免的伴隨著能源的大量消耗,為了實(shí)現(xiàn)雙碳目標(biāo),尋找能夠提高能源利用率和減少碳排放的節(jié)能途徑刻不容緩。相變材料優(yōu)良的蓄放熱特性和高的儲能密度[1-2],使其具有巨大的儲能應(yīng)用潛力,被廣泛應(yīng)用于建筑、太陽能和冷鏈運(yùn)輸?shù)阮I(lǐng)域[3-8],但多數(shù)相變材料受到腐蝕性、泄露和易緩慢氧化老化等缺點(diǎn)的限制[9-13]。相變材料微膠囊的出現(xiàn)克服了上述的問題,可以降低外部環(huán)境對相變材料的影響,增加材料的換熱面積,避免材料對容器的腐蝕,并且可控制相變材料體積變化[14]。基于上述優(yōu)點(diǎn),相變微膠囊在紡織、建筑節(jié)能和電池?zé)峁芾硐到y(tǒng)等領(lǐng)域日益受到重視[15]。常用的微膠囊制備方法包括原位聚合法、乳液聚合法和懸浮聚合法等,其中原位聚合法是制備微膠囊的主要方法[16]。包裹相變材料的壁材主要包括脲醛樹脂、密胺樹脂和聚甲基丙烯酸甲酯等材料,其中脲醛樹脂通常被認(rèn)為是原位聚合法制備相變材料的良好候選材料[17-18]。
已有研究表明,在原位聚合法制備微膠囊的過程中,乳化劑種類、乳化劑HLB值和初始pH值等對微膠囊的形貌和分散性有直接影響[19]。王信剛等[20]利用原位聚合法制備了脲醛樹脂包覆癸酸的相變微膠囊,并設(shè)計正交試驗用于分析制備條件對微膠囊的影響。實(shí)驗結(jié)果表明,水溶性表面活性乳化劑OP-10與油溶性表面活性乳化劑Span80組成復(fù)配乳化劑,其中疏水基的范德華力和親水基的極性引力使彼此能形成強(qiáng)的協(xié)同作用,提高了癸酸乳液的穩(wěn)定性,能夠合成形貌良好、分散性優(yōu)異的癸酸微膠囊。王書穎等[21]采用原位聚合法制備了十四烷-密胺樹脂微膠囊,在制備過程中,將三聚氰胺分3次加入制備預(yù)聚體溶液,制備了微膠囊。結(jié)果表明,pH值在3.5~3.7范圍內(nèi),制備的微膠囊球形完整,粒徑分布均勻,殘余甲醛含量低。張小英等[22]同樣采用原位聚合法制備了十四烷-密胺樹脂微膠囊,實(shí)驗研究了復(fù)配乳化劑的HLB值對微膠囊形貌和分散性的影響,認(rèn)為乳化劑的HLB值為12.03時,微膠囊表面形貌、熱性能和分散性良好。
本文通過原位聚合法制備了一系列十四烷-脲醛樹脂微膠囊。研究了制備微膠囊過程中乳化劑種類、HLB值、預(yù)聚體水量和初始pH值對微膠囊形貌、粒徑和包覆率的影響,并得到了最優(yōu)的微膠囊制備條件。
1 實(shí) 驗
1.1 原材料與實(shí)驗儀器
尿素(Urea),購于無錫市晶科化工有限公司;甲醛溶液(Formaldehyde,37%(質(zhì)量分?jǐn)?shù))),購于上海聯(lián)式化工試劑有限公司;三乙醇胺,購于天津市恒興化學(xué)試劑制造有限公司;十四烷(Tet)、間苯二酚和冰乙酸購于上海阿拉丁生化科技股份有限公司;Span80和Tween80,購于上海山浦化工有限公司;正辛醇,購于天津博迪化工股份有限公司;氯化銨,購于天津市致遠(yuǎn)化學(xué)試劑有限公司;去離子水,自制。實(shí)驗過程中使用的儀器見表1。
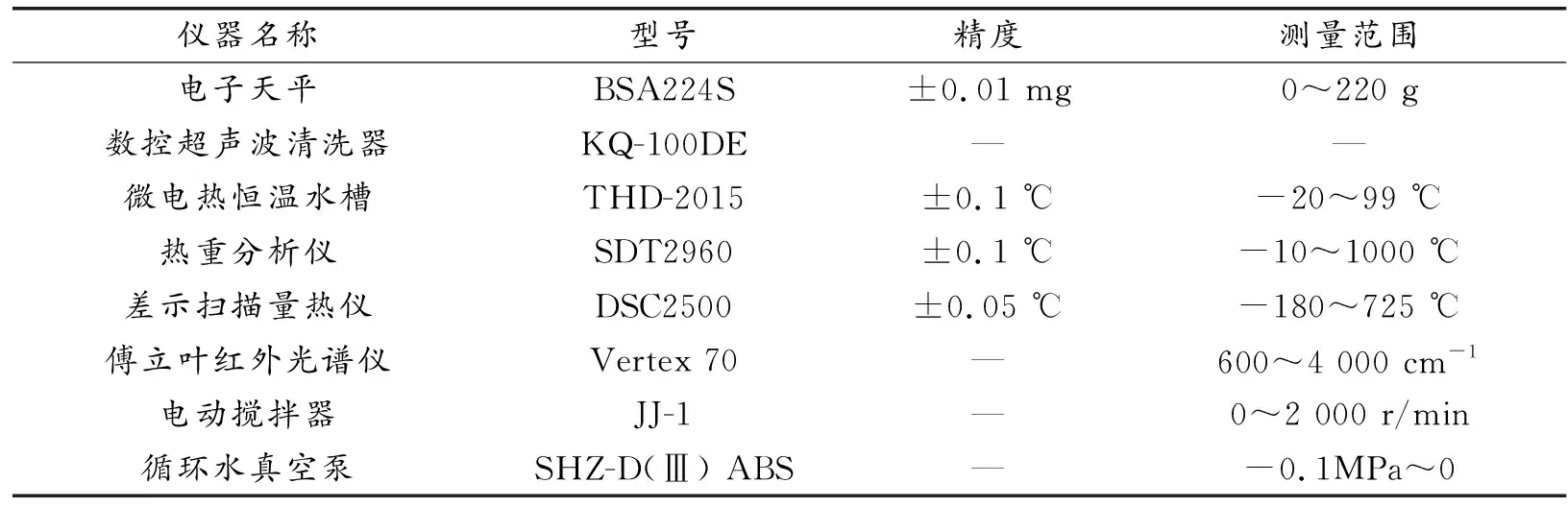
表1 實(shí)驗儀器Table 1 Experimental equipment information
1.2 微膠囊的制備
微膠囊的制備過程如圖1所示。首先將5.8 g尿素,11.5 g甲醛溶液(37%(質(zhì)量分?jǐn)?shù)))和一定量的去離子水添加至燒杯內(nèi)混合,添加2滴三乙醇胺使得溶液pH值為8~9。將上述溶液添加至三口燒瓶中,采用電動攪拌器以500 r/min的速度攪拌,用65 ℃水浴加熱60 min得到脲醛樹脂預(yù)聚體;隨后將5 g十四烷、一定量乳化劑(Span80-Tween80、OP-10-SDBS或SDBS)和50 g去離子水組成混合液。將混合液轉(zhuǎn)移到三口燒瓶中,在常溫下采用電動攪拌器以2 000 r/min的速度攪拌30 min得到十四烷乳液,然后將制備好的脲醛樹脂預(yù)聚體緩慢的滴加至乳液中,再稱取0.58 g間苯二酚和0.58 g NH4Cl溶解于10 g去離子水中,隨后緩慢添加5%(質(zhì)量分?jǐn)?shù))的冰乙酸至溶液中,調(diào)節(jié)體系的pH值(3.0、3.5和4.0)。在縮聚階段,電動攪拌器的轉(zhuǎn)速為300 r/min,反應(yīng)溫度由室溫以1 ℃/min的速度升高至60 ℃,并保持4.5 h。最后采用乙醇和去離子水沖洗抽濾3次得到十四烷-脲醛樹脂微膠囊,再采用烘干機(jī)5 h烘干后回收。實(shí)驗研究了乳化劑種類、HLB值、預(yù)聚體水量和初始pH值對微膠囊制備的影響,實(shí)驗體系見表2。
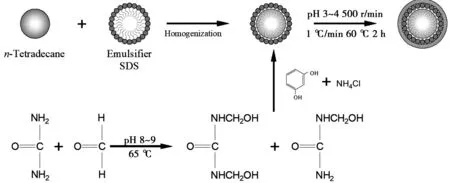
圖1 十四烷微膠囊的合成過程圖Fig 1 Synthesis process diagram oftetradecane microcapsules
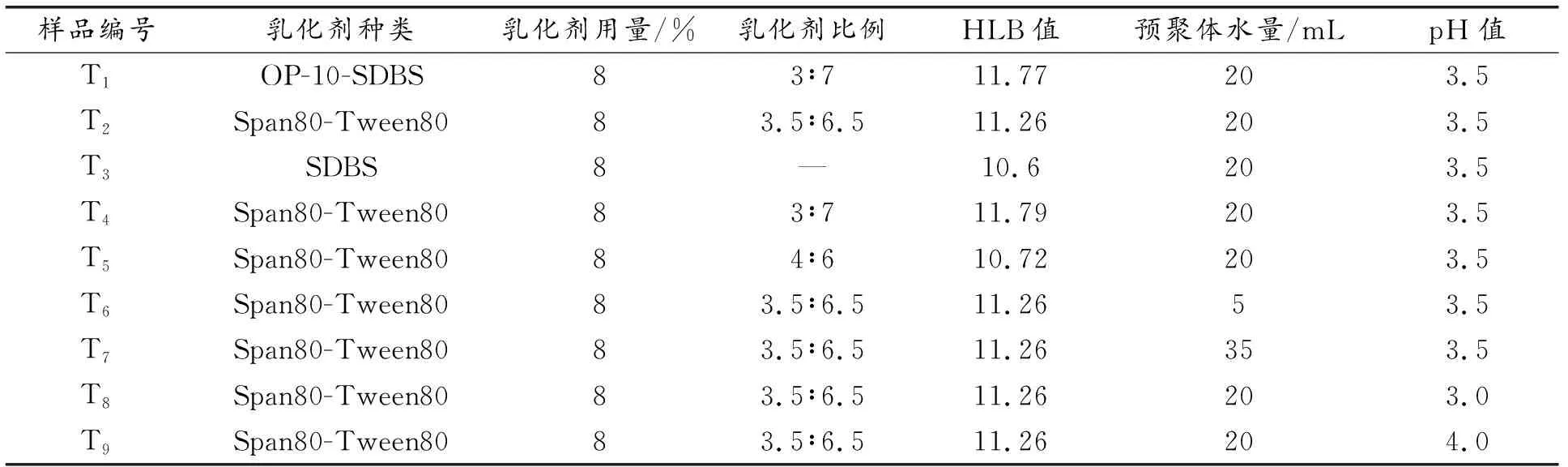
表2 原位聚合法制備十四烷微膠囊的合成參數(shù)表Table 2 Synthetic parameter table oftetradecane microcapsules prepared by in-situ polymerization
1.3 材料性能表征
采用美國FEI公司生產(chǎn)的Quanta FEG 250掃描電子顯微鏡觀察微膠囊的形態(tài),其加速電壓為10 kV。微膠囊的相變熱和相變溫度采用美國TA公司生產(chǎn)的TA差示掃描量熱儀DSC2500測試,在氮氛圍下控制測試溫度為-20 ℃至20 ℃,升溫速率為5 ℃/min。微膠囊粒徑由馬爾文電位粒度儀測量。利用Vertex 70傅里葉紅外光譜儀測量了材料的譜圖,該設(shè)備的掃描范圍為4 000~600 cm-1,分辨率為4 cm-1,樣品掃描次數(shù)為16次。測試時,取約1 mg樣品粉末,加入稀釋劑溴化鉀(約150 mg)研磨均勻,并進(jìn)行壓片處理。采用美國TA公司生產(chǎn)的差熱熱重聯(lián)用儀SDT2960對微膠囊進(jìn)行了TG熱重測試,在氮氛圍下測試溫度為室溫至600 ℃,升溫速率為10 ℃/min。
2 結(jié)果與討論
2.1 乳化劑種類對微膠囊的影響
在制備微膠囊的過程中,乳化劑種類對乳液的穩(wěn)定性和微膠囊的合成有較大大的影響。徐慧等[23]研究了不同乳化劑對十四烷乳液的穩(wěn)定性,實(shí)驗表明HLB值在11左右的乳化劑更易得到穩(wěn)定的乳液。本文選取OP-10-SDBS、Span80-Tween80和SDBS 3種HLB值在11左右的乳化劑制備了穩(wěn)定的十四烷乳液,然而在制備微膠囊的過程中得到了截然不同的結(jié)果,如圖2所示。從SEM圖像上可以看出,樣品T1以O(shè)P-10-SDBS為乳化劑制備得到微膠囊出現(xiàn)大量破損,粒徑不均一,微膠囊表面是由聚脲微顆粒組成,表面存在漏洞。出現(xiàn)這一現(xiàn)象的原因是由于乳化劑OP-10在體系升溫過程中無法保持乳液的穩(wěn)定,產(chǎn)生一定的熟化和破乳現(xiàn)象,隨著反應(yīng)時間的推移,在體系酸度不斷下降的影響下,低分子量脲醛樹脂和脲醛樹脂初級粒子的沉積受到擾亂,出現(xiàn)了聚合后的脲醛樹脂微顆粒再次聚集的現(xiàn)象,導(dǎo)致微膠囊結(jié)構(gòu)不穩(wěn)定和破損。樣品T2以Span80-Tween80為乳化劑制備得到的微膠囊表面光滑,分散性好,熱穩(wěn)定性優(yōu)異。與OP-10-SDBS復(fù)合乳化劑體系相比,Span80的HLB值為4.3易溶于油相,Tween80的HLB值為15易溶于水相,Span80-Tween80乳化劑制備得到的乳液在體系酸度逐步下降的同時,能夠保持乳液的穩(wěn)定性,阻止熟化和破乳現(xiàn)象的發(fā)生,使得低分子量脲醛樹脂能夠穩(wěn)定的形成脲醛樹脂納米殼層,脲醛樹脂初級顆粒能逐步穩(wěn)定的沉積,從而形成表面為光滑,穩(wěn)定性優(yōu)異的微膠囊。樣品T3采用SDBS陰離子乳化劑制備微膠囊,表現(xiàn)出了破裂、團(tuán)聚和大量脲醛樹脂微顆粒的雜亂粘附,且微膠囊的壁材較薄,穩(wěn)定性較差。以SDBS為乳化劑的乳液在體系反應(yīng)后期由于酸度降低導(dǎo)致乳液穩(wěn)定性變差,在完成脲醛樹脂微膠囊納米殼層的生成后便無法進(jìn)一步吸附脲醛樹脂初級顆粒生成壁材,導(dǎo)致脲醛樹脂初級顆粒生成團(tuán)聚,并雜亂的粘附在微膠囊表面。因此,在后面的工作中選用Span80-Tween80復(fù)配乳化劑制備微膠囊。

圖2 不同乳化劑種類條件下制備的微膠囊SEM圖像Fig 2 SEM images of microcapsules synthesized under different emulsifier types
2.2 乳化劑HLB值對微膠囊的影響
在微膠囊合成過程中,HLB值也是影響其形貌和分散性的重要因素,因此在選定Span80-Tween80復(fù)配乳化劑的情況下,對體系HLB值進(jìn)行微調(diào),并制備了樣品T2、T4和T5,實(shí)驗結(jié)果如圖3。樣品T4中Span80-Tween80比例為3∶7(HLB值為11.79),微膠囊表面粗糙,且凹凸不平,粒徑也不均勻,含有大量細(xì)小的脲醛樹脂顆粒,同時伴隨著團(tuán)聚現(xiàn)象發(fā)生。樣品T2中Span80-Tween80比例為3.5∶6.5(HLB值為11.26),微膠囊表面光滑,分散性好,粒徑分布較平均。樣品T5的復(fù)配表面活性劑Span80-Tween80比例為4∶6時(HLB值為10.72),微膠囊表現(xiàn)出大量的團(tuán)聚,表面形態(tài)和比例3∶7的情況類似。Span80-Tween80比例在3∶7至4∶6范圍內(nèi),十四烷乳液都能夠保持穩(wěn)定。實(shí)驗表明,在縮聚反應(yīng)的過程中,與十四烷微膠囊反應(yīng)體系最匹配的HLB值為11.26,當(dāng)HLB值高于此值時,體系水相內(nèi)Tween-80含量增加從而形成膠束,導(dǎo)致大量細(xì)小的脲醛樹脂顆粒形成,進(jìn)一步影響其分散性;當(dāng)HLB值低于此值時,體系水相內(nèi)Tween-80含量降低,直接導(dǎo)致脲醛樹脂初級顆粒的分散性降低,導(dǎo)致微膠囊的團(tuán)聚。

圖3 Span80-Tween80體系制備的微膠囊SEM圖像Fig 3 SEM images of microcapsules synthesized with Span80-Tween80
2.3 預(yù)聚體水量對微膠囊的影響
兩步法制備微膠囊在縮聚過程需要將預(yù)聚體緩慢滴加至體系中。因此,對于剛剛制備完成的乳液和整個反應(yīng)體系,預(yù)聚體的添加量影響著縮聚過程中乳液的穩(wěn)定性。當(dāng)制備預(yù)聚體的去離子水量為20 mL時,樣品T2表現(xiàn)出優(yōu)異的分散性和形貌,如圖4(b)。然而,當(dāng)樣品T6的預(yù)聚體制備過程中去離子水的添加量為5 mL時,雖然在緩慢滴加至體系中時會減少對乳液穩(wěn)定性的影響,但是整個反應(yīng)體系的水量減少了15 mL,直接導(dǎo)致水相中脲醛樹脂初級顆粒濃度升高,進(jìn)而導(dǎo)致了脲醛樹脂微顆粒大量生成、脲醛樹脂初級顆粒的雜亂沉積和嚴(yán)重的團(tuán)聚現(xiàn)象,如圖4(a)。當(dāng)制備預(yù)聚體的去離子水量為35 mL時,在滴加過程中會導(dǎo)致乳液穩(wěn)定性受到影響,因此樣品T7產(chǎn)生了微膠囊粒徑不均和輕微團(tuán)聚的問題。隨著體系水量的增加,水相中脲醛樹脂初級顆粒和乳化劑的濃度降低,同時伴隨著乳液的不穩(wěn)定,導(dǎo)致了脲醛樹脂初級顆粒在生成的壁材出現(xiàn)粗糙、輕微破損和輕微團(tuán)聚的現(xiàn)象,如圖4(c)。

圖4 預(yù)聚體水量對微膠囊影響Fig 4 The influence ofprepolymer water volume on microcapsules
2.4 初始pH值對微膠囊的影響
樣品T2、T8和T9的SEM圖像如圖5所示,不同初始pH值下微膠囊的形貌和分散性差異較大。在樣品T8的縮聚過程中,初始pH值為3.0會引起反應(yīng)速率無法得到有效地控制,導(dǎo)致了脲醛樹脂初級顆粒出現(xiàn)大量無法控制的自聚,進(jìn)而影響沉積過程使得微膠囊的形貌,無法達(dá)到規(guī)則的球形,同時出現(xiàn)大量的團(tuán)聚,但仍然能夠完成對十四烷的包裹,如圖5(a)。縮聚過程初始pH值為4.0雖然能夠減緩脲醛樹脂初級顆粒的聚集和沉積過程,但是緩慢的反應(yīng)速度配合不斷下降的體系酸度導(dǎo)致的乳液熟化,樣品T9產(chǎn)生了微膠囊粒徑不均一和團(tuán)聚的現(xiàn)象。同時,緩慢的反應(yīng)速率使得聚合過程的微膠囊長期處于不穩(wěn)定的狀態(tài),伴隨著攪拌器產(chǎn)生的剪切應(yīng)力,導(dǎo)致微膠囊表面出現(xiàn)孔洞和破裂,出現(xiàn)芯材十四烷的泄露,如圖5(c)。當(dāng)體系初始pH值調(diào)節(jié)至3.5時,微膠囊壁材的生成速度與乳液保持穩(wěn)定的時段契合,因此樣品T2表面光滑,具有優(yōu)異的分散性。因而本文選取縮聚過程初始pH值為3.5作為優(yōu)選的制備條件。

圖5 不同pH值制備微膠囊的SEM圖像Fig 5 SEM images of microcapsules synthesized with different pH values
根據(jù)上述實(shí)驗結(jié)果,我們得到了最佳的制備條件,即樣品T2:乳化劑采用Span80和Tween80復(fù)配乳化劑,Span80和Tween80的質(zhì)量比為3.5∶6.5,預(yù)聚體制備過程中去離子水添加量為20 mL,并且將pH值調(diào)節(jié)至3.5,則可制備表面形貌和分散性均十分優(yōu)異的高性能微膠囊。
2.5 微膠囊粒徑分析
十四烷微膠囊的粒徑分布如圖6,測試結(jié)果表明微膠囊的粒徑分布主要集中在1~20 μm范圍內(nèi),其平均粒徑為8.4 μm。約有5%的微膠囊粒徑在40~200 μm范圍內(nèi),該現(xiàn)象是由于在微膠囊制備過程中縮聚階段,大部分乳液在保持穩(wěn)定的狀態(tài)下形成微膠囊,但有少部分乳液發(fā)生熟化導(dǎo)致粒徑增加,另有部分微膠囊在遠(yuǎn)離攪拌葉片的區(qū)域發(fā)生輕微團(tuán)聚,因此導(dǎo)致了部分微膠囊粒徑較大。
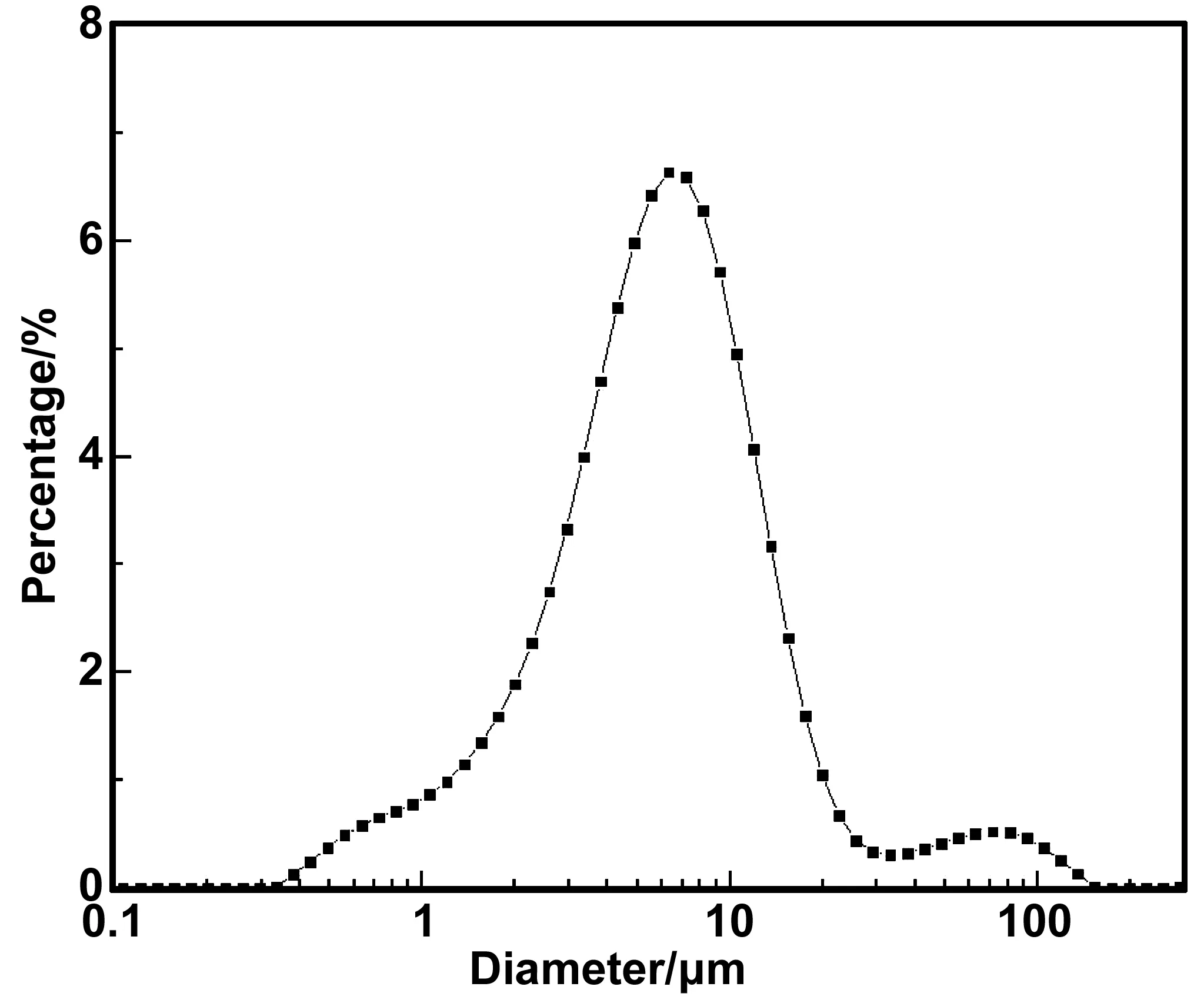
圖6 十四烷微膠囊的粒徑Fig 6 The particle size oftetradecane microcapsules
2.6 微膠囊相變儲能能力分析
十四烷微膠囊的DSC測試結(jié)果如圖7所示。微膠囊的熔融起始溫度為3.05 ℃,峰值溫度為5.27 ℃,其相變焓為59.65 J/g,微膠囊的凝固起始溫度為0.88 ℃,峰值溫度為0.65 ℃,其相變焓為60.21 J/g。微膠囊的相變溫度符合低溫蓄冷潛熱型功能流體等領(lǐng)域的實(shí)際工程應(yīng)用范圍,將其應(yīng)用于冷凍水循環(huán)系統(tǒng)中,可以提高系統(tǒng)的蓄冷能力,減小系統(tǒng)充液量和管徑,提高能源利用率。
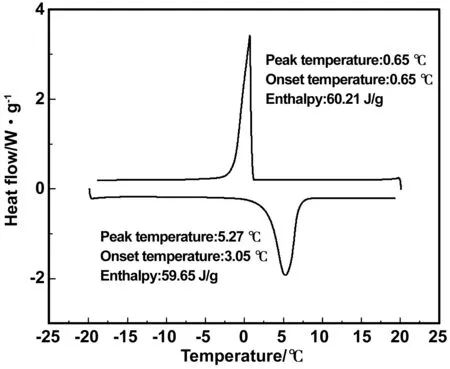
圖7 十四烷微膠囊的DSC圖Fig 7 DSC picture oftetradecane microcapsules
2.7 紅外光譜分析
芯材十四烷、脲醛樹脂和微膠囊的紅外光譜如圖8所示。3種物質(zhì)的光譜圖像在特征圖上匹配十分緊密,如CH3在2 926 cm-1處的不對稱伸縮振動峰和CH2在2 853 cm-1處的對稱伸縮振動峰。O—H基團(tuán)在3 600~3 200 cm-1范圍內(nèi)呈現(xiàn)寬吸收帶。在1 000 cm處C—O的伸縮振動峰,1 641 cm-1處C=O的伸縮振動吸收峰和1 559 cm-1處的C—N的伸縮振動吸收峰都密切匹配。結(jié)果表明,十四烷被成功包裹,在合成過程中沒有新物質(zhì)生成。
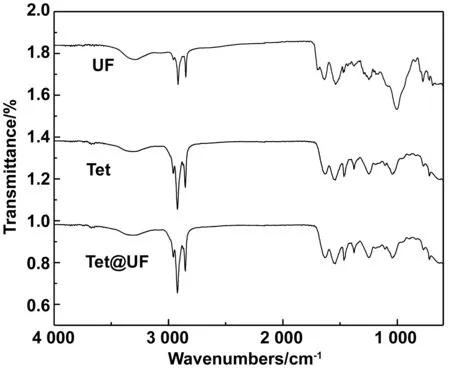
圖8 十四烷、脲醛樹脂和十四烷微膠囊的紅外光譜圖Fig 8 Infrared spectra of tetradecane, urea-formaldehyde resin and tetradecane microcapsules
2.8 微膠囊的熱穩(wěn)定性
分別對芯材十四烷、脲醛樹脂和微膠囊進(jìn)行TG測試。從圖9中可以看出,十四烷在86 ℃左右開始分解,在147 ℃完全分解;脲醛樹脂在133 ℃左右開始分解,在465 ℃完全分解;十四烷-脲醛樹脂微膠囊在0~72 ℃范圍內(nèi)沒有失重,于72~115 ℃時,隨著微膠囊所含水分的不斷蒸發(fā),質(zhì)量損失為5%。在溫度從115 ℃升高至162 ℃時,脲醛樹脂壁材開始破裂,芯材十四烷不斷分解,在453 ℃左右壁材脲醛樹脂完全分解。TG實(shí)驗表明,十四烷-脲醛樹脂微膠囊在蓄冷應(yīng)用溫度范圍內(nèi)熱穩(wěn)定性良好,能夠滿足其在潛熱型功能流體和冷鏈物流等低溫儲能領(lǐng)域的應(yīng)用。
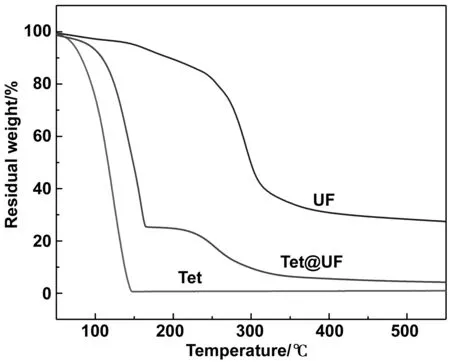
圖9 十四烷、脲醛樹脂和十四烷微膠囊的熱重曲線Fig 9 TG curves oftetradecane, urea-formaldehyde resin and tetradecane microcapsules
3 結(jié) 論
(1) 乳化劑的種類及其HLB值影響微膠囊的制備,采用Span80-Tween80復(fù)配的乳化劑能夠制備得到形貌優(yōu)異,球形完整的微膠囊。在相同乳化劑用量的情況下,乳化劑HLB值的變化會導(dǎo)致表面粗糙和團(tuán)聚的現(xiàn)象,當(dāng)Span80和Tween80的質(zhì)量比為3.5∶6.5(HLB=11.26)時,微膠囊的形貌光滑,分散性優(yōu)異。
(2) 在兩步法制備微膠囊的過程中,預(yù)聚體水量會影響乳液的穩(wěn)定性和預(yù)聚體初級顆粒的濃度,進(jìn)而導(dǎo)致微膠囊出現(xiàn)表面粗糙、輕微破損和輕微團(tuán)聚的現(xiàn)象。
(3) 體系縮聚過程中的初始pH值直接影響微膠囊的形貌和分散性,較低的初始pH值會引起反應(yīng)速率無法得到有效地控制,導(dǎo)致微膠囊的團(tuán)聚;在較高的初始pH值情況下,緩慢的反應(yīng)速度以及不斷下降的體系酸度產(chǎn)生了微膠囊粒徑不均一和團(tuán)聚的現(xiàn)象。pH值控制在3.5,制備的微膠囊形貌優(yōu)異,分散性好。
(4) 采用原位聚合法制備的芯壁比為1∶2的微膠囊平均粒徑為8.4 μm,相變溫度和相變潛熱分別為3.05 ℃和60 J/g。
猜你喜歡
商品與質(zhì)量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀(jì)智能(數(shù)學(xué)備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛(wèi)生(2015年12期)2015-11-10 05:13:40
現(xiàn)代企業(yè)(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
