原發性皮膚CD30陽性間變性大細胞淋巴瘤1例
2022-08-04 06:23:02蘇群英黃炳臣吳雪銘王興枝子龍喜帶
臨床與實驗病理學雜志 2022年6期
蘇群英,黃炳臣,吳雪銘,王 娟,王興枝子,龍喜帶
患者女性,63歲,因發現右前臂、頸背部病灶,逐漸增大伴局部潰瘍、滲液半年余。2020年7月20日患者于右江民族醫學院附屬醫院進行治療,自訴半年前無意中發現右前臂、頸背部有“米粒”大小的結節性淡紅色病灶,曾多次于我院皮膚科行“封閉注射”治療,但病灶日漸隆起增大,范圍明顯增寬,并伴局部潰爛、滲液,自感疼痛、瘙癢,影響日常睡眠及生活。實驗室檢查無明顯改變,缺乏特異性特征。
病理檢查眼觀:右前臂可見一大小5 cm×4 cm淡紅色病灶,呈半球狀明顯隆起皮面,質硬,稍壓痛,邊界清,侵犯皮膚并與之粘連,活動度可(圖1)。頸背部可見一大小6 cm×4 cm病灶,呈淡紅色,半球形,質硬,邊界清,侵犯皮膚并與之黏連,活動度可,中央可見局部潰爛、流膿,周圍皮膚紅腫(圖2)。鏡檢:腫瘤細胞彌漫性浸潤至真皮及皮下組織,瘤細胞大,有明顯異型性,核仁明顯,胞質豐富,核分裂象易見。免疫表型:CD30(圖3)、CD4、CD2、MUM-1(IRF4)均陽性,ALK、CD20、CD56均陰性,Ki-67增殖指數約80%。原位雜交檢測:EBER陰性;FISH檢測:IRF4基因有分離信號(圖4)。
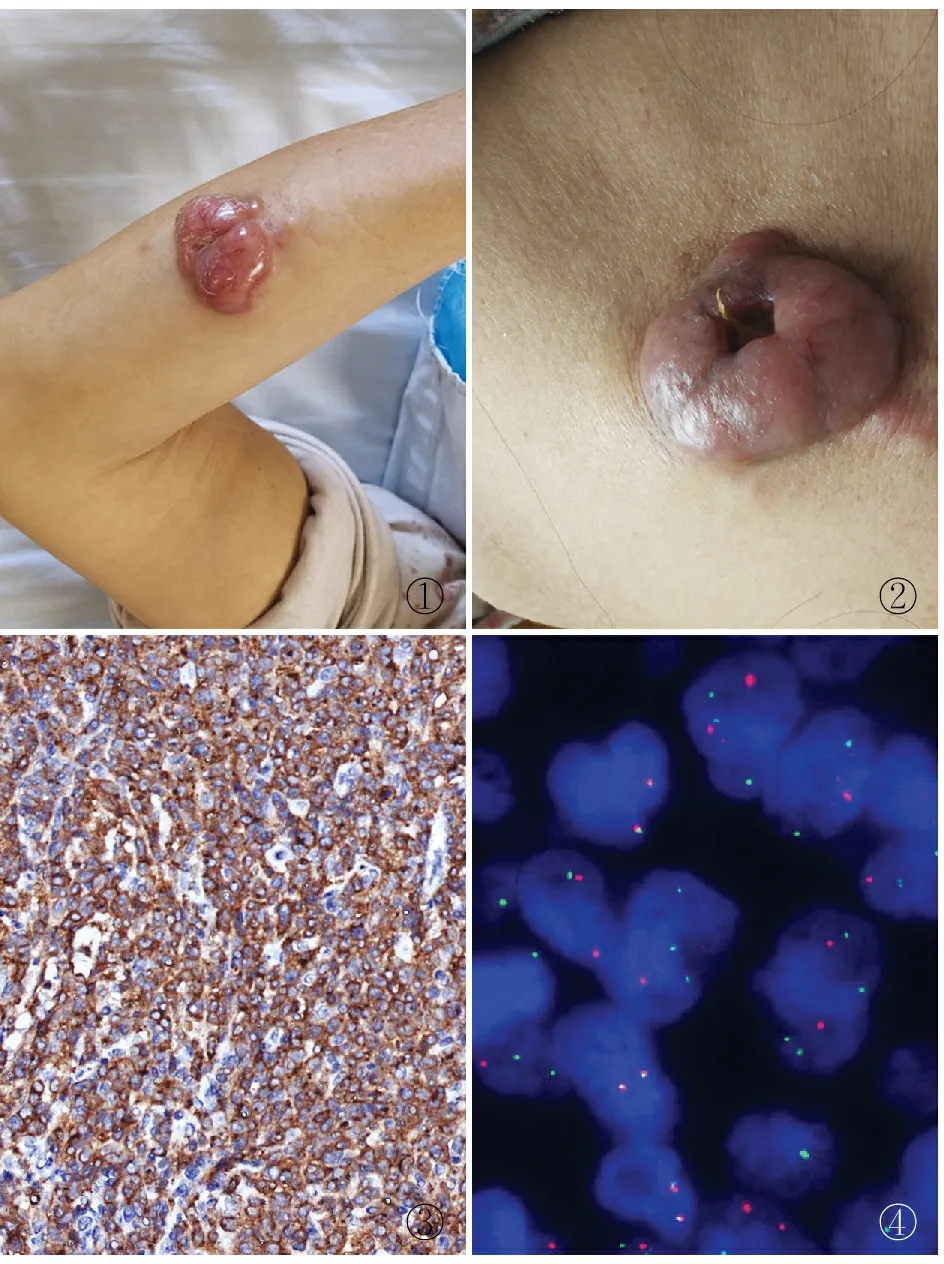
圖1 右前臂腫物:皮膚表面有一大小5 cm×4 cm淡紅色病灶,呈半球狀明顯隆起 圖2 頸背部腫物:皮膚表面見一大小6 cm×4 cm病灶,侵犯皮膚并與之黏連,中央可見局部潰爛、流膿 圖3 腫瘤細胞胞膜CD30陽性,EnVision法 圖4 FISH檢測示IRF4基因有分離信號
病理診斷:(右前臂、頸背部)原發性皮膚CD30陽性間變性大細胞淋巴瘤。
討論間變性大細胞淋巴瘤(anaplastic large cell lym-phoma, ALCL)于1985年被Stein等首次命名,CD30強陽性,是一種具有獨特的臨床表現、免疫表型及遺傳學特點的淋巴增生性疾病,屬于非霍奇金細胞淋巴瘤亞型,約占3%。根據WHO(2016)造血與淋巴組織腫瘤分類將ALCL分為4類:系統型ALCL(ALK+ALCL、ALK-ALCL)、原發性皮膚間變性大細胞淋巴瘤(primary cutaneous anaplastic large cell lymphoma, PC-ALCL)和乳房植入物相關的間變性大細胞淋巴瘤(breast implant associated anaplastic large cell lymphoma, BIAALCL)[1-3]。
PC-ALCL多見于老年人,平均發病年齡60歲,男性多于女性,兒童偶見,通常為紅色至紫紅色的孤立性腫瘤或結節簇,直徑大于2 cm,腫物生長迅速且常伴潰瘍,約20%的患者存在多中心性,患者常無臨床癥狀。組織學特征為大淋巴樣組織的包繞,瘤細胞胞核呈圓形、卵圓形、腎形、不規則的馬蹄形核,核仁嗜酸性且細胞質豐富,細胞浸潤延伸到真皮或皮下脂肪層,而除了存在潰瘍情況下表皮不受累及。免疫組化標記至少有75%的大細胞以膜狀或高爾基體模式表達CD30;T細胞抗原表達CD4,但CD2、CD3和CD5不同程度表達丟失。
PC-ALCL主要與淋巴瘤丘疹樣病(lymphomatoid papulosis, LyP)、系統型ALCL累及皮膚鑒別。原發性皮膚CD30陽性T細胞淋巴組織增殖性疾病是一組譜系疾病,包括LyP和PC-ALCL[3]。根據WHO(2016)淋巴造血組織疾病分類可將LyP分為六個組織學亞型(A~F亞型),尤其是C型表現為CD30陽性的大T淋巴樣細胞結節內浸潤,多數無炎性細胞,類似于ALCL。因此,在形態學上難以與PC-ALCL鑒別,需結合臨床表現以及分子檢測進行鑒別。系統型ALCL的繼發性皮膚病變和PC-ALCL的組織學類似,但兩者預后差異較大,PC-ALCL為局限性皮膚病變、缺乏淋巴結腫大、年齡通常大于30歲,而75%的系統型ALCL有B癥狀,特別是有發熱癥狀,ALK+ALCL發病年齡小于30歲的患者,應結合臨床資料加以鑒別。此外,還需與臨床表現和組織學形態相近的黑色素瘤、低分化癌、蟲咬皮炎等進行鑒別。
研究表明,ALK-ALCL和PC-ALCL均可檢測DUSP22和TP63的重排。DUSP22和TP63的重排分別占PC-ALCL的30%和5%,DUSP22重排的ALCL患者臨床預后良好,而TP63重排的ALCL患者臨床預后較差[4-5]。PC-ALCL預后好,10%~42%的病變可自發消退,5、10年生存率均超過90%,但PC-ALCL易復發(40%)。低劑量甲氨蝶呤是廣泛或皮外受累的一線全身治療藥物首選,聯合化療藥物方案以CHOP方案為主[6]。本例患者術后1個月腫瘤復發,給予聯合化療藥物CHOP方案治療,目前患者情況良好。
