C-S-H納米晶核對礦物摻合料復合膠凝材料水化性能的影響研究
2022-08-08 07:36:26劉鳳東楊飛華呂民望王發洲
硅酸鹽通報 2022年7期
關鍵詞:體系
王 坤,劉鳳東,楊飛華,呂民望,楊 露,王發洲
(1.武漢理工大學硅酸鹽建筑材料國家重點實驗室,武漢 430070;2.武漢理工大學材料科學與工程學院,武漢 430070; 3.固廢資源化利用與節能建材國家重點實驗室,北京 100041;4.北京建筑材料科學研究總院有限公司,北京 100041)
0 引 言
礦物摻合料以其經濟性、豐富性和對水泥性能改善的優異性被廣泛用于水泥混凝土中,已成為高性能混凝土施工與應用不可缺少的第六組分[1-4]。鋼渣、礦渣、硅灰、粉煤灰等礦物摻合料是工業生產過程中產生的固體廢棄物,因其在較高的溫度環境下生成,具有潛在的火山灰性和膠凝性,可作為輔助膠凝材料取代部分水泥用量,不僅降低了水泥在生產和使用中所帶來的碳排放壓力[5-7],還有利于水泥混凝土的長期性能[8-9]。Radlinski等[10]提出采用輔助膠凝材料來減少水泥用量,起到合理利用工業廢棄物,改善水泥混凝土的使用性能[11]和實現水泥行業的可持續發展等多重好處。
當礦物摻合料作為輔助膠凝材料加入到水泥體系后,由于其早期的反應活性不如微集料效應明顯,因此對水泥漿體的早期強度發展具有一定的抑制作用[12-14]。C-S-H晶核是一種納米成核劑,引入到水泥體系中可以降低部分水化產物的成核勢壘,加速水化歷程,促進C-S-H凝膠和鈣礬石等主要水化產物的生成。石磊等[15]將C-S-H晶核摻入水泥體系中研究其水化速度和物理性能,結果表明晶核的引入可以促進水泥的水化,有效縮短水泥凝結時間,改善早期強度。因此,在水泥-礦物摻合料體系中摻雜C-S-H納米晶核可加速水泥-礦物摻合料體系的早期水化反應速度,促進水化產物的快速生成,有效彌補礦物摻合料所帶來的早期缺陷,改善其早期活性和后期性能。
本文通過熱力學計算闡述了C-S-H晶核對水泥水化產物成核勢壘的影響,并采用微量熱儀、pH計和電導率測試儀等對摻合料復合膠凝材料稀溶液進行檢測,測試其離子溶出濃度與沉積物物相結構,探究了C-S-H納米凝膠晶核對復合膠凝材料體系水化活性和水化歷程的影響規律,為大摻量礦物摻合料復合膠凝材料設計與理論研究提供參考。
1 實 驗
1.1 原材料
硅酸鹽水泥為福建金牛水泥廠生產的P·Ⅰ 52.5水泥;礦物摻合料為福建金牛水泥廠的Ⅱ級粉煤灰(fly ash, FA)和粒化高爐礦渣微粉(ground granulated blast furnace slag, GGBFS);C-S-H納米晶核為江蘇蘇博特新材料公司生產的納米C-S-H晶核,固含量為14.1%(質量分數)。水泥和礦物摻合料的化學成分如表1所示,水泥的礦物組成和率值如表2所示。
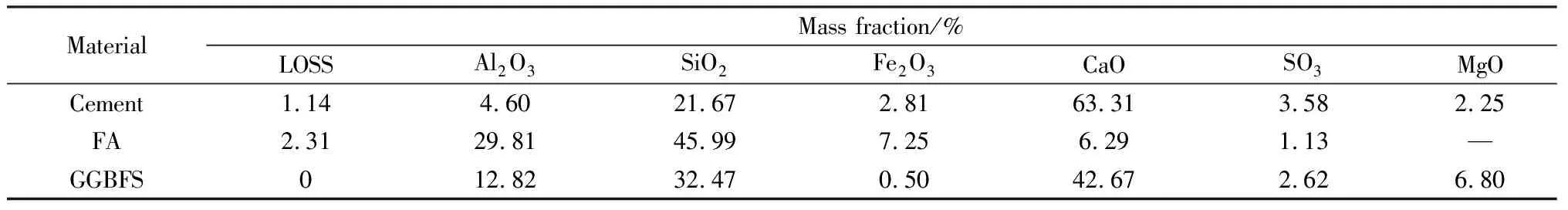
表1 水泥和礦物摻合料的主要化學成分Table 1 Main chemical composition of cement and mineral admixtures

表2 水泥的礦物組成和率值Table 2 Mineral composition and rate value of cement
1.2 試驗方法
采用FA和GGBFS分別取代30%(質量分數)的水泥用量,采用外摻法加入膠凝材料用量2%(質量分數的C-S-H晶核形成對照組,水膠比(W/B)取為40,配制稀溶液,在室溫下不斷攪拌,加速其水化歷程,具體配比如表3所示。
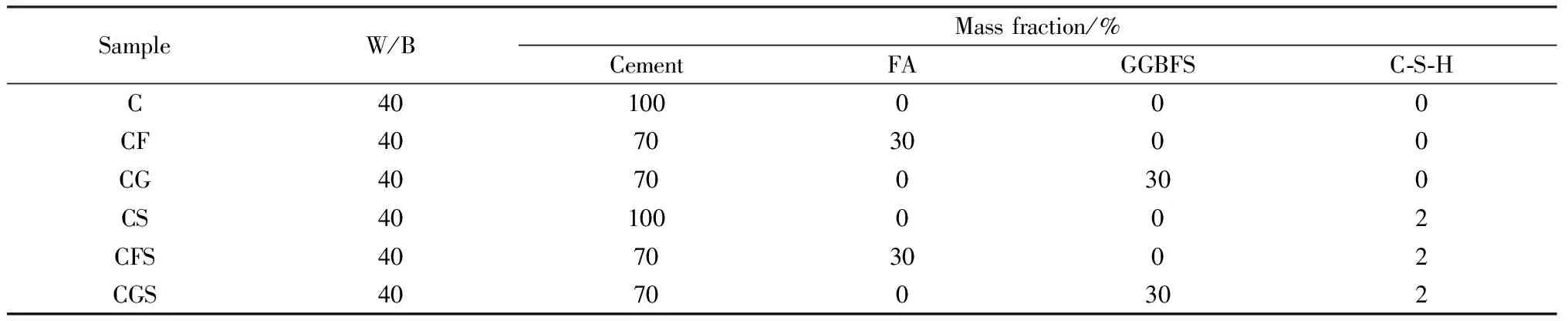
表3 稀溶液配比Table 3 Proportion of dilute solution
1.2.1 pH和電導率測試
按照試驗設計配比稱取15 g粉料和600 g去離子水于三口燒瓶中,加入磁力攪拌子持續攪拌,燒瓶兩側分別插入pH和電導率儀器探頭,每隔3 min記錄一組數據,持續監測24 h。
1.2.2 等離子體發射光譜(ICP)
按照試驗設計配比稱取1 g粉料和40 mL去離子水于樣品管中,每個配比配制7個樣品,分別對應0.5 h、1 h、2 h、4 h、6 h、12 h和24 h七個不同水化時間節點,置于恒溫震蕩箱中不斷震蕩。在對應時間節點取出樣品離心,取酸化后的濾液測試其離子含量。
1.2.3 沉積物物相結構分析
對不同水化時間節點的離心沉積物進行XRD和TG-DTG測試,研究其水化產物微結構。
1.2.4 水化熱分析
按照配比稱取10 g膠凝材料和5 g去離子水配制漿體,采用一致攪拌的方法以消除攪拌對水化作用的影響[16]。由于水的比熱容大約是膠凝材料的5倍,故取參比水為7 g,采用八通道微量熱儀(Thermometrics TAM air)測試各樣品水化1 d的放熱速率和總放熱量。
2 C-S-H納米晶核的作用機理
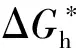
(1)
(2)
由熱力學公式可知,均勻成核勢壘與非均勻成核勢壘之間僅相差一個接觸角系數f(θ),當接觸角θ越小時,f(θ)越小,非均勻成核下的成核勢壘越小,水化產物越容易結晶長大。
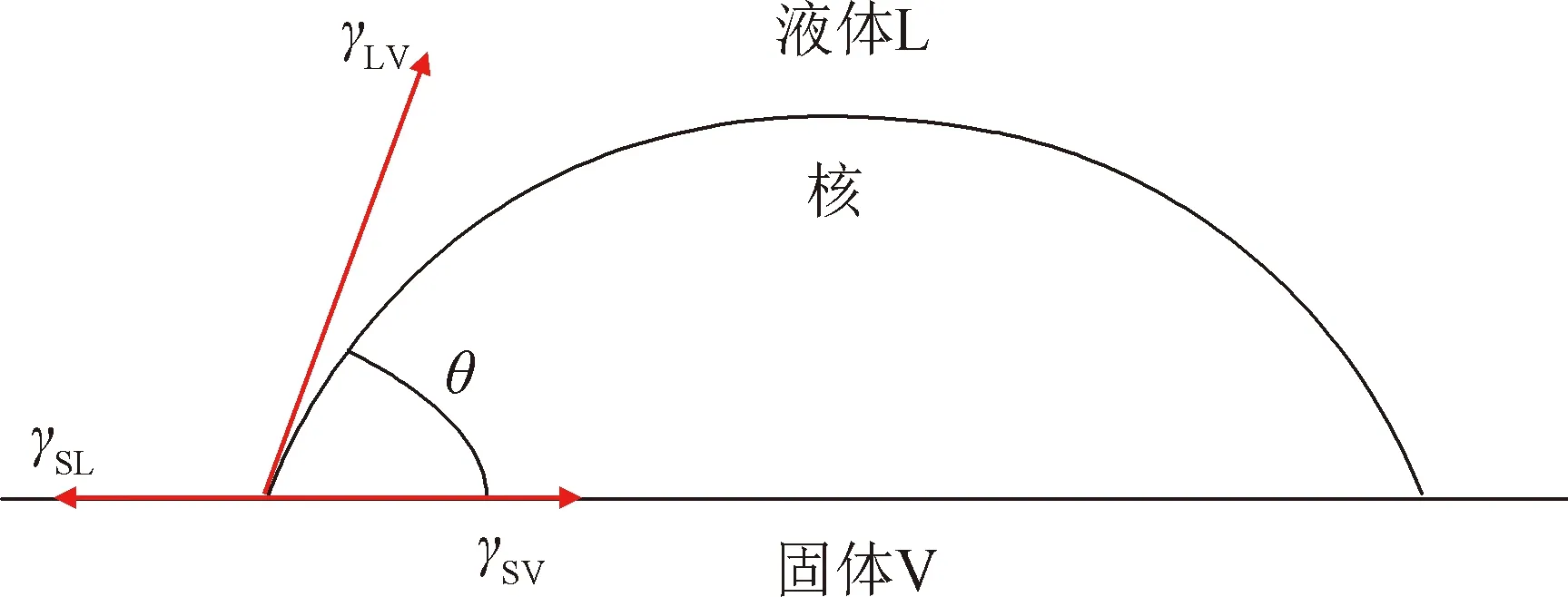
圖1 液體-固體界面非均勻成核示意圖Fig.1 Schematic diagram of non-uniform nucleation at the liquid-solid interface
圖1為液-固界面非均勻成核的示意圖。young方程為:
γSV=γSL+γLV·cosθ
(3)
式中:γSV、γSL、γLV分別表示固氣、固液和液氣界面的界面張力;θ為平衡接觸角。
由式(3)可知,接觸角由兩相間界面能決定,與各相表面能有關。王政等[19]對氫氧化鈣和水化硅酸鈣在晶核表面結晶進行熱力學計算,其結果表明氫氧化鈣和C-S-H凝膠在人工合成晶核表面結晶的接觸角分別為89°和0°,氫氧化鈣的成核勢壘降低約50%,C-S-H凝膠則可以在晶核表面直接成核。
晶核作為成核劑引入到液相中,新相成核會受到影響,成核優先于晶核表面寄生,使得氫氧化鈣(CH)和C-S-H凝膠等水化產物成核勢壘大幅降低,復合膠凝材料體系水化活性得以提升。
3 結果與討論
3.1 水化量熱分析
圖2為復合膠凝材料體系24 h水化放熱速率和累計放熱圖。如圖2所示,無晶核下純水泥漿體的誘導期較復合膠凝材料體系短,這是礦物摻合料早期水化活性并不顯著,其對水泥所起的稀釋作用使得水化歷程往后偏移。當摻入2%的晶核后,各體系誘導期大幅縮短,放熱速率和累計放熱均有提升,出現有明顯的尖銳峰。摻入晶核的體系在5.5 h左右就達到峰值,比無晶核體系時間縮短了一半。CFS和CGS組的放熱速率達到了6.2 mW/g,1 d的累計放熱也達到了200 J/g左右,超越了C組,說明在晶核的作用下,水泥體系中硅酸鹽相活性得到激發,早期參與水化反應的能力被大幅加強,水化歷程被縮短,促進了AFt(三硫型硫鋁酸鈣)向AFm(單硫型硫鋁酸鈣)的轉化,使得復合膠凝材料體系的水化活性接近純水泥體系。因此,晶核的引入能改善礦物摻合料所帶來的早期水化放熱低和水化能力不足等缺陷。
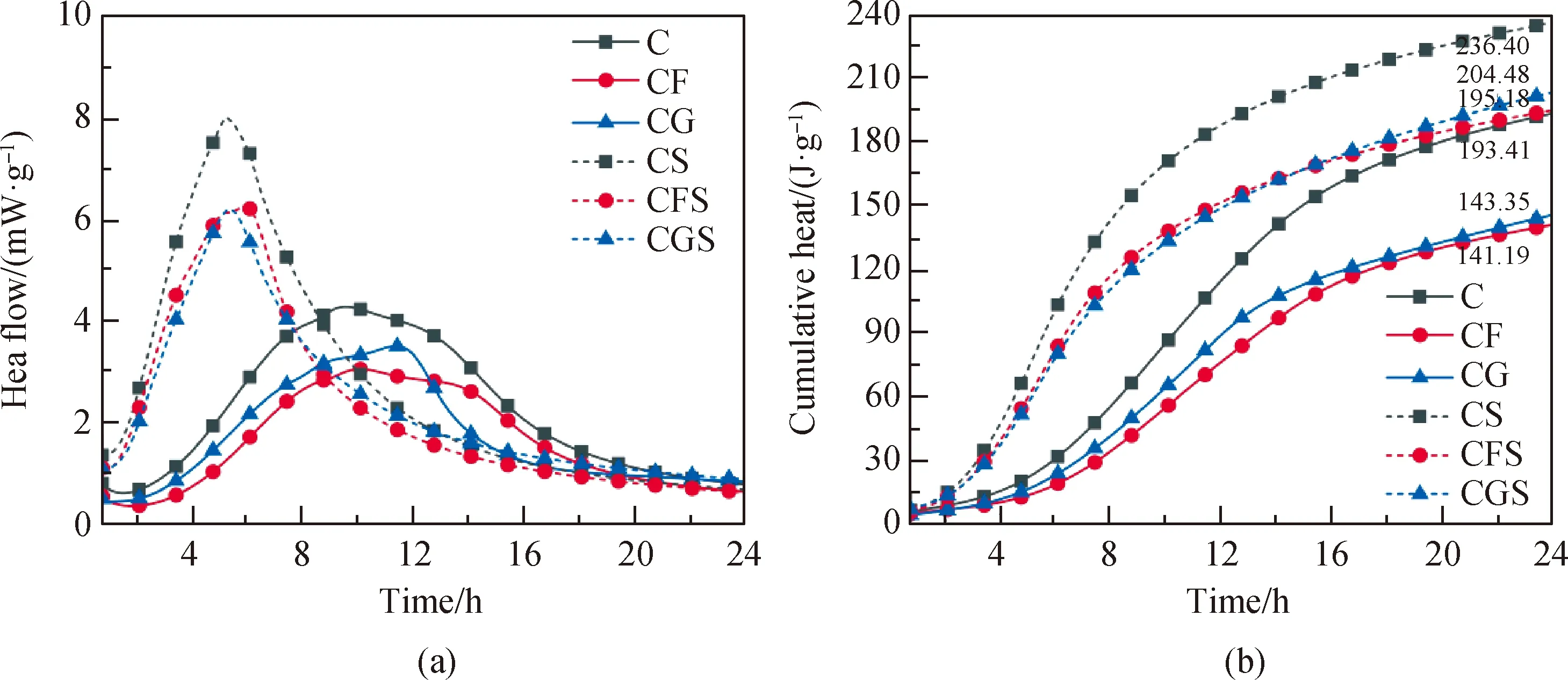
圖2 復合膠凝材料體系24 h水化放熱速率和累計放熱Fig.2 Hydration heat release rate and cumulative heat release diagram of composite cementitious material system
3.2 pH值和電導率分析
通過對大水膠比復合膠凝材料稀溶液pH值和電導率的24 h連續檢測,觀察其在水泥水化過程中的發展趨勢,并探究礦物摻合料對水泥水化以及晶核對復合膠凝材料體系水化過程的影響,試驗結果如圖3所示。圖3(a)為24 h pH值測試圖,溶液中pH值主要是受到OH-和堿離子濃度的影響,當FA和GGBFS部分取代水泥后,體系中Ca2+、K+、Na+和OH-等離子濃度隨著水泥用量減少而降低,使得體系pH值均部分降低,由純水泥的13.2降至12.8左右,其中FA的降低程度要大于GGBFS,這是由于礦粉的早期溶出活性要優于粉煤灰。當引入晶核后,純水泥體系和水泥-礦粉體系pH值小幅降低,說明晶核可以加速[SiO4]4-和Ca2+的消耗,促進水化產物的生成,導致孔隙溶液中pH值有所降低;而對于水泥-粉煤灰體系,晶核的引入使得pH值有所上升,這是因為粉煤灰中含有大量的鋁,其溶出會以離子形態吸附在硅酸鹽相表面溶痕處及活性位點處,抑制硅酸鹽相的進一步溶解反應,但會導致過多的堿性離子富集在孔溶液中,對體系堿度產生影響。
圖3(b)、(c)為稀溶液24 h電導率測試結果圖。由圖可知,電導率的發展可分為三個階段:(1)增長期,各礦相快速溶解,溶液中的離子含量迅速增加,電導率不斷增大至峰值,此階段溶解速率大于沉積速率;(2)下降期,隨著溶液中離子含量的不斷增長,水泥水化產物快速沉積生成,使得離子含量有所降低,電導率開始下降,此階段沉積速率大于溶解速率;(3)平穩期,隨著礦相溶解和水化產物沉積生成的不斷進行,水泥的水化進入穩定期,此時溶解速率和沉積速率相當,電導率幾乎保持不變。如圖3(b)所示,純水泥在持續攪拌8.6 h左右電導率就達到峰值。當引入礦物摻合料后,峰值降低,出現的時間推遲5 h,說明礦物摻合料的引入降低水泥用量,對體系的溶解和沉積起到了稀釋作用,使得電導率峰值和平穩期的穩定值均有所降低;當向復合膠凝材料體系中加入2%的C-S-H納米晶核后,各體系電導率峰值均較早出現且有顯著提升,使得復合膠凝材料體系電導率接近純水泥體系,如圖3(c)所示,說明晶核可以加速復合膠凝材料體系的溶出-沉積速率,縮短水化歷程,大幅減弱礦物摻合料取代水泥所帶來的活性降低和水化慢等負面效果。
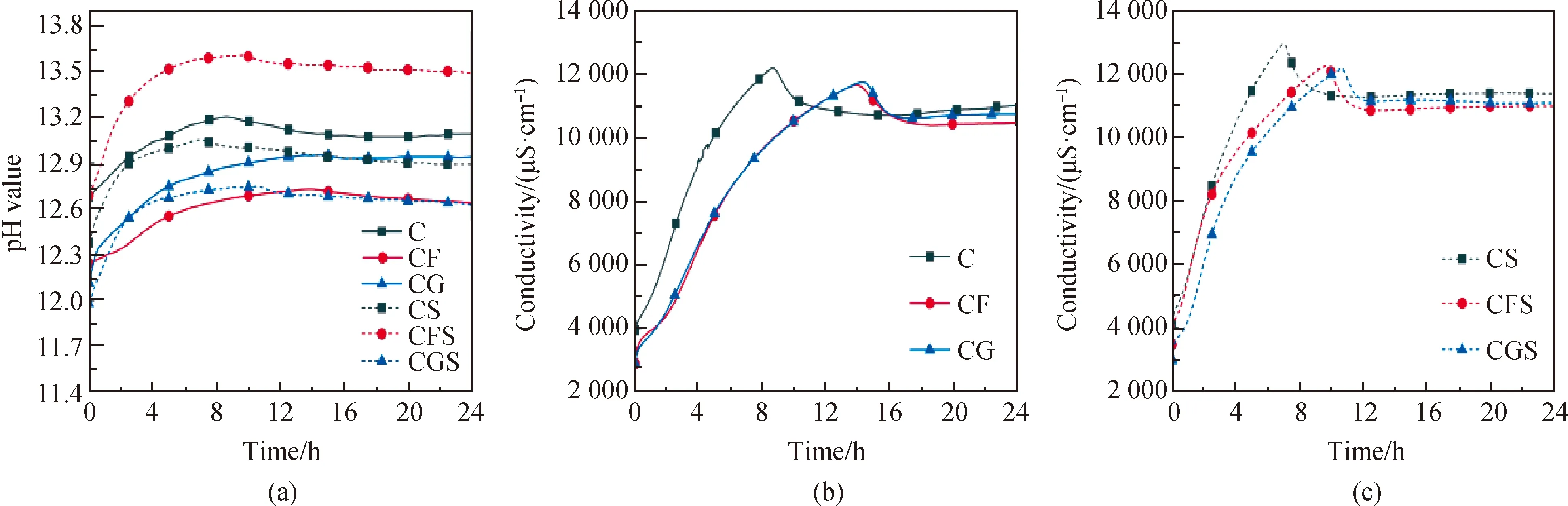
圖3 復合膠凝材料體系稀溶液中pH值和電導率Fig.3 pH value and conductivity in the dilute solution of composite cementitious material system
3.3 ICP分析
為了進一步研究晶核對復合膠凝材料體系水化歷程的影響,對不同水化時間節點(水化0.5 h、1 h、2 h、4 h、6 h、12 h、24 h)的稀溶液進行震蕩離心,取上層清液酸化進行ICP檢測,研究晶核對復合膠凝材料體系稀溶液中鈣硅鋁鐵等離子含量變化的影響,其結果如圖4所示。由圖可知,在水化早期,無論摻加晶核與否,膠凝材料體系中Ca元素濃度均顯著高于Si、Al和Fe的濃度,原因在于熟料礦物中各化學成分并非一致溶出,其中Ca離子更易溶出。在引入C-S-H納米晶核后鈣離子溶出速度加快,而含有硅、鋁元素的離子溶出速率被阻礙,說明晶核的引入促進了水泥中硅酸鹽相的溶解水化,使得溶液中CH含量大幅度上升,飽和點提前到來;此外晶核提供了C-S-H凝膠的成核位點,降低了成核勢壘,使得溶液中的含硅元素的離子被快速吸收,水化產物C-S-H凝膠大量生成。此外,C-S-H納米晶核能夠使熟料礦物C3S等溶出的[SiO4]4-迅速聚合成C-S-H凝膠而使得其在溶液中的濃度相比于未摻晶核樣品更低,這一過程進一步促進了C3S礦物的解體,產生更多的鈣離子,證明了納米C-S-H晶核對水泥熟料礦物水化的進一步促進作用。
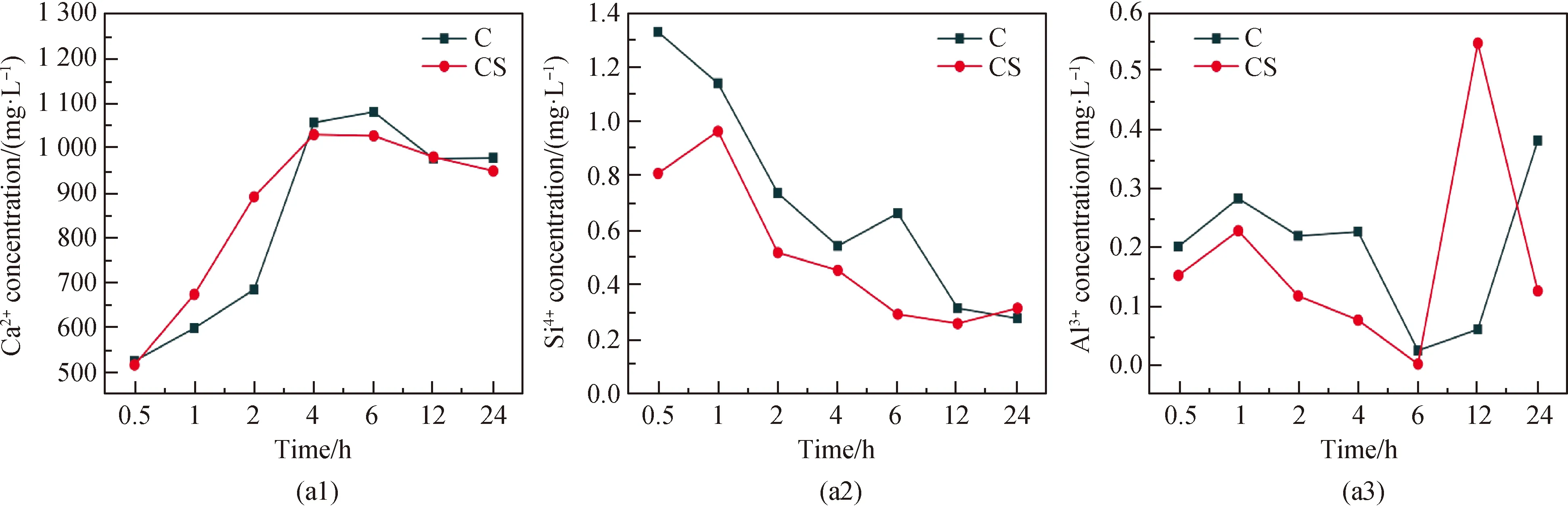
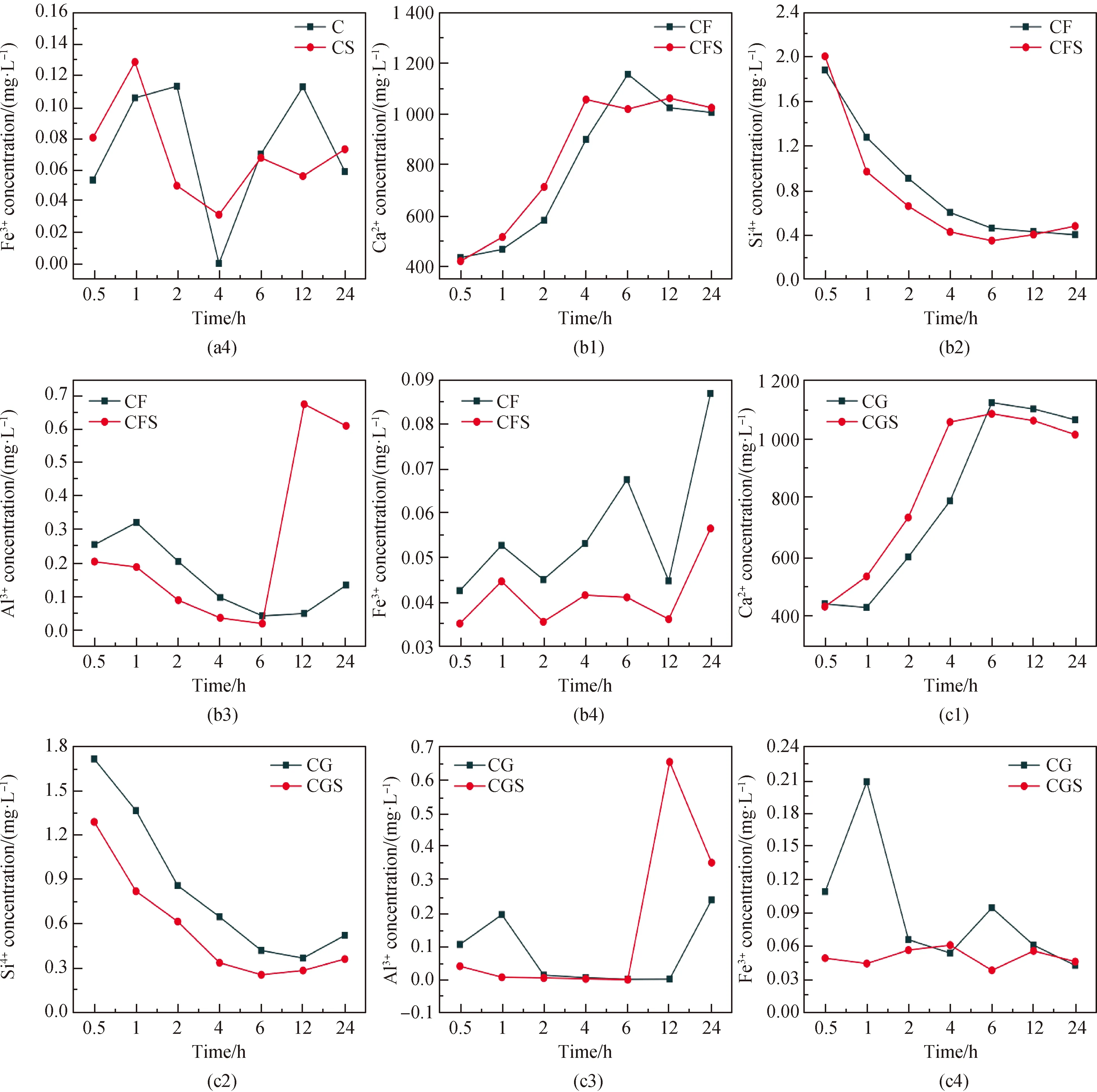
圖4 復合膠凝材料體系稀溶液中元素濃度Fig.4 Development of ion concentration of composite cementitious material system
3.4 X射線衍射分析
圖5為不同水化時間節點下各復合膠凝材料體系水化產物XRD譜。如圖所示,當使用礦物摻合料取代部分水泥用量時,體系早期C3S和AFt的衍射峰無明顯變化。CF組在水化6 h后出現一個尖銳的CH峰,而在CFS組中卻消失了,這是因為FA早期活性不如其微集料效應明顯,對溶液中的鈣離子消耗速度緩慢,CH逐漸飽和析出。但隨著時間延長,FA的火山灰性得到激發,鈣離子消耗速度增大,導致析出的CH晶體又會重新溶解在不飽和溶液中。當引入晶核后,硅酸鹽相溶解迅速,溶液堿度增大,FA活性激發時間提前,在6 h前就會有大量CH析出,后又重新溶解,整個過程都比無晶核體系迅速。對于礦粉復合膠凝材料體系,礦粉的火山灰性較強,對于溶液中大量溶解的鈣離子具有一定的消耗作用,使得溶液中CH難以飽和結晶析出,因此難以捕捉到CH的強衍射峰。對于晶核復合膠凝材料體系,各階段的C3S和AFt峰值均有所降低,說明晶核通過吸收[SiO4]4-促進了體系中硅酸鹽相的溶解水化,使得AFt轉變成AFm時間提前。同時,在晶核和大水膠比持續攪拌的作用下,各體系水化歷程被大幅縮減,溶解產生的CH快速與礦物摻合料發生二次水化,因此晶核能夠激發復合膠凝材料體系的活性,促進了水化反應的發生和產物的生成。
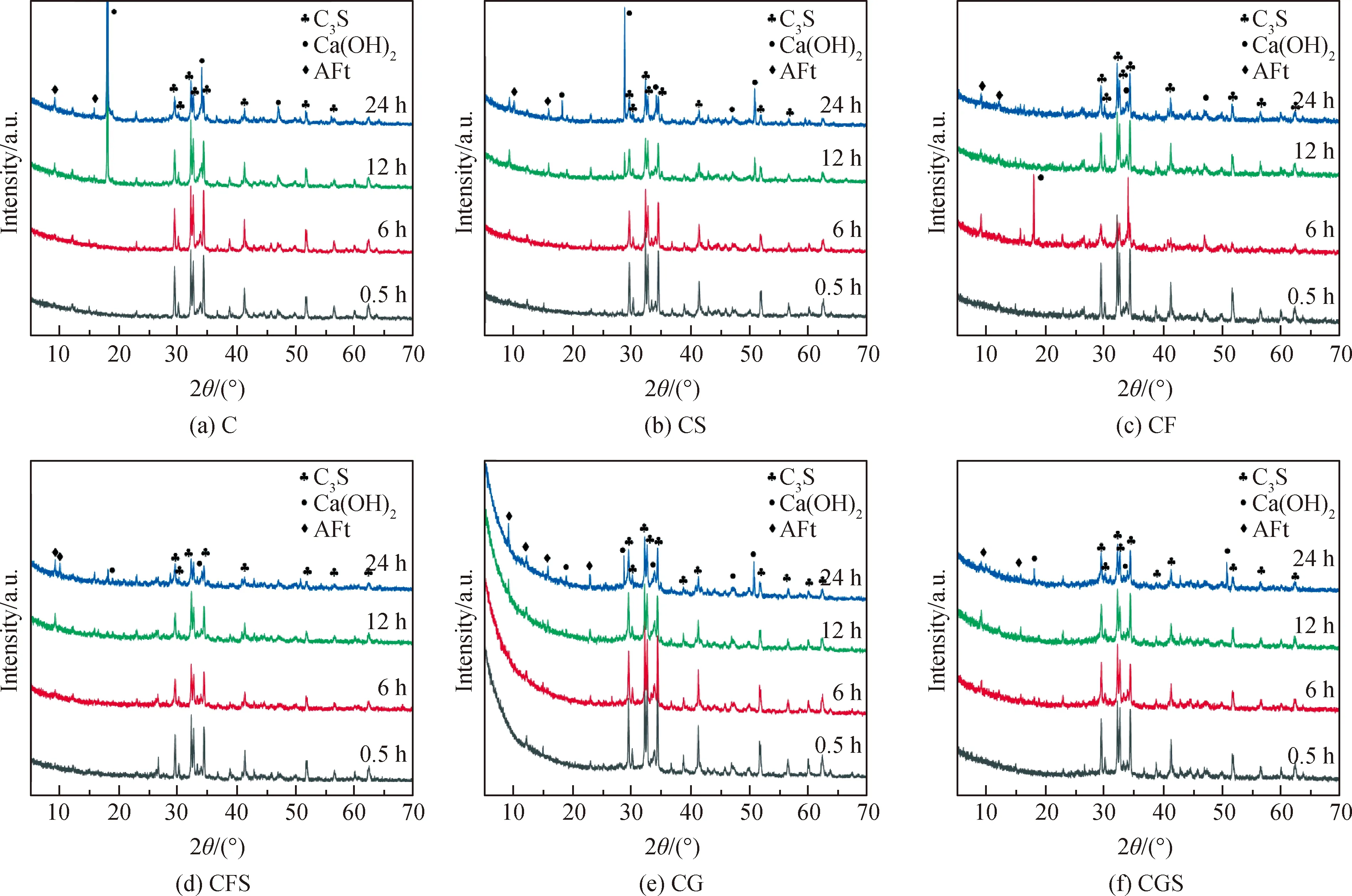
圖5 不同水化時間節點下各復合膠凝材料體系水化產物XRD譜Fig.5 XRD patterns of hydration products of each composite cementitious material system under different hydration time
3.5 TG-DTG分析
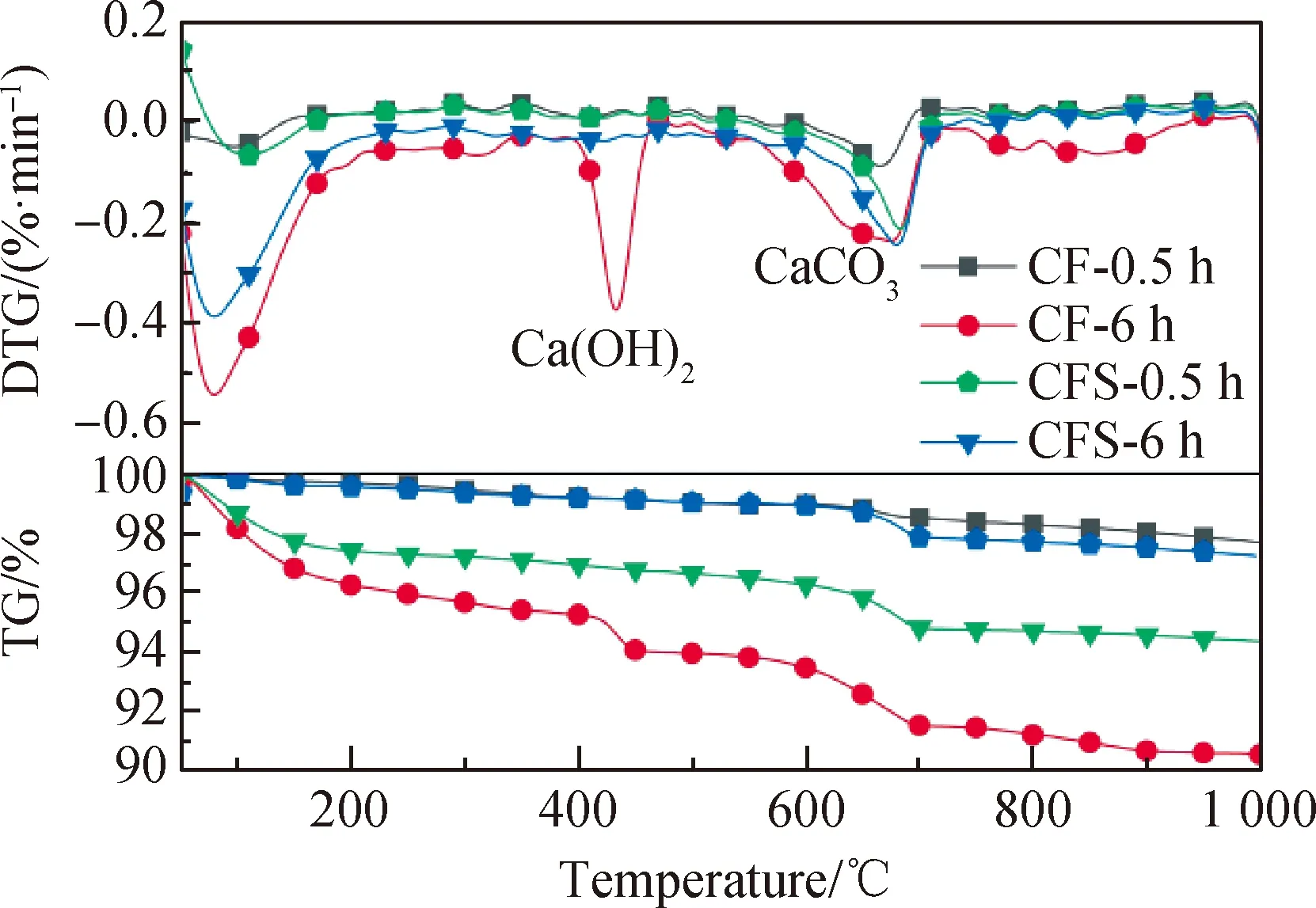
圖6 粉煤灰復合膠凝材料體系沉積相TG-DTG曲線Fig.6 TG-DTG curves of sedimentary phase of fly ash composite cementitious material system
為進一步探究晶核對復合膠凝材料體系早期水化活性的影響,優選了粉煤灰復合膠凝材料體系,測試在水化30 min和6 h 沉積相TG-DTG曲線,結果如圖6所示。由TG-DTG測試結果可以看出,晶核在水化30 min時便可以產生促進效果,使得沉積物中CH和凝膠量增多;當水化6 h后,無晶核體系沉積物中出現了CH峰,說明FA早期活性較低,與水泥水化產生的鈣離子結合速度緩慢,導致富余的鈣離子飽和析出。CFS組水化6 h后未出現明顯的CH峰,說明晶核促進了礦物摻合料的二次水化,使得溶液中的鈣離子消耗速度加快,難以達到飽和,已析出的CH會重新溶解在溶液中。因此晶核可以促進礦物摻合料的火山灰活性使得復合膠凝材料體系水化歷程大幅縮短,早期離子溶出,遷移及沉積性能得到改善。
4 結 論
(1)礦物摻合料復合膠凝材料體系放熱緩慢,水化活性和離子溶出程度低,礦物摻合料對水泥顆粒的稀釋作用使得體系水化歷程往后偏移。
(2)納米晶核作為成核劑可提高復合膠凝材料離子溶出程度,降低成核勢壘,促進氫氧化鈣和水化硅酸鈣等水化產物的生成,可以彌補復合膠凝材料體系與水泥體系之間的活性差距。
(3)C-S-H納米晶核促進硅酸鹽相溶解與Ca2+溶出,同時給復合膠凝材料體系水化提供一個高堿性環境,極大的提升了體系的水化能力。晶核作為納米成核劑,可為大摻量低活性礦物摻合料復合膠凝材料體系設計與理論研究提供參考。
猜你喜歡
商品與質量(2021年43期)2022-01-18 05:31:22
杭州(2020年23期)2021-01-11 00:54:42
新世紀智能(數學備考)(2020年11期)2021-01-04 00:38:16
中國外匯(2019年17期)2019-11-16 09:31:14
中國衛生(2015年12期)2015-11-10 05:13:40
現代企業(2015年1期)2015-02-28 18:43:18
汽車零部件(2014年5期)2014-11-11 12:24:28
新高考·高一物理(2014年1期)2014-09-18 01:26:07
浙江人大(2014年1期)2014-03-20 16:19:53
終身教育研究(2012年4期)2012-03-25 10:41:11
