
一體化電極電催化二氧化碳還原研究進展
2022-08-10 03:41:02宋雨珂謝文富邵明飛
物理化學學報 2022年6期
關鍵詞:催化劑
宋雨珂,謝文富,邵明飛
北京化工大學化學學院,化工資源有效利用國家重點實驗室,北京 100029
1 引言
工業革命以來,化石燃料(煤、石油、天然氣等)的過度使用造成大氣中的CO2含量逐年上升,已嚴重超過自然界的碳循環限度,引起了一系列環境問題,如氣溫升高和氣候劇變1-4。CO2轉化技術,不僅可以有效緩解CO2劇增所帶來的環境問題,還可以制備高附加值的燃料或化學品5,6。迄今為止,CO2轉化技術可劃分為四類:生物化學法、熱化學法、光化學法和電化學法7-12。其中,電化學CO2轉化技術憑借反應條件相對溫和、轉化效率高和反應步驟可控的優點引起了研究者們的廣泛關注。此外,太陽能、風能等清潔能源可以為電化學CO2轉化技術提供電能,為其大規模的工業應用提供了廣闊的前景11,13,14。
電化學CO2還原反應(E-CO2RR)是一個包含多質子和電子轉移的復雜過程(圖1a)15,如何實現對于特定產物的可控催化成為當前E-CO2RR的研究重點。為了加快E-CO2RR的速率,同時使得反應選擇性地生成目標產物,研究者們開發了眾多高活性、高選擇性的E-CO2RR催化劑。通過精細調控催化劑的電子結構,如構筑缺陷16-18、空位19-21、合金22,23、特定晶界24-27和晶面28-30,可以實現對中間產物吸脫附過程的有效控制,進而促進催化活性和選擇性。然而,通過改變電子結構調控催化劑本征活性時,不同中間產物(如*CO、*COH、*CHOH和*CHO等)在催化劑表面的吸附行為存在線性關聯,加強其中一種中間產物的吸附,必然伴隨著其他中間產物的吸附增強,難以實現E-CO2RR整體性能增強(圖1b)31,32。除了電子結構調控,表界面微環境調控也可以顯著影響催化劑的E-CO2RR性能。例如,催化劑的微納結構精細控制可以改變其表面的CO2濃度、催化劑-電解液界面的pH以及反應物和產物傳質過程等,進而有望改善E-CO2RR催化活性、選擇性和穩定性等。此外,該類特性往往不受線性關聯的制約,在E-CO2RR領域具有重要的研究價值33。
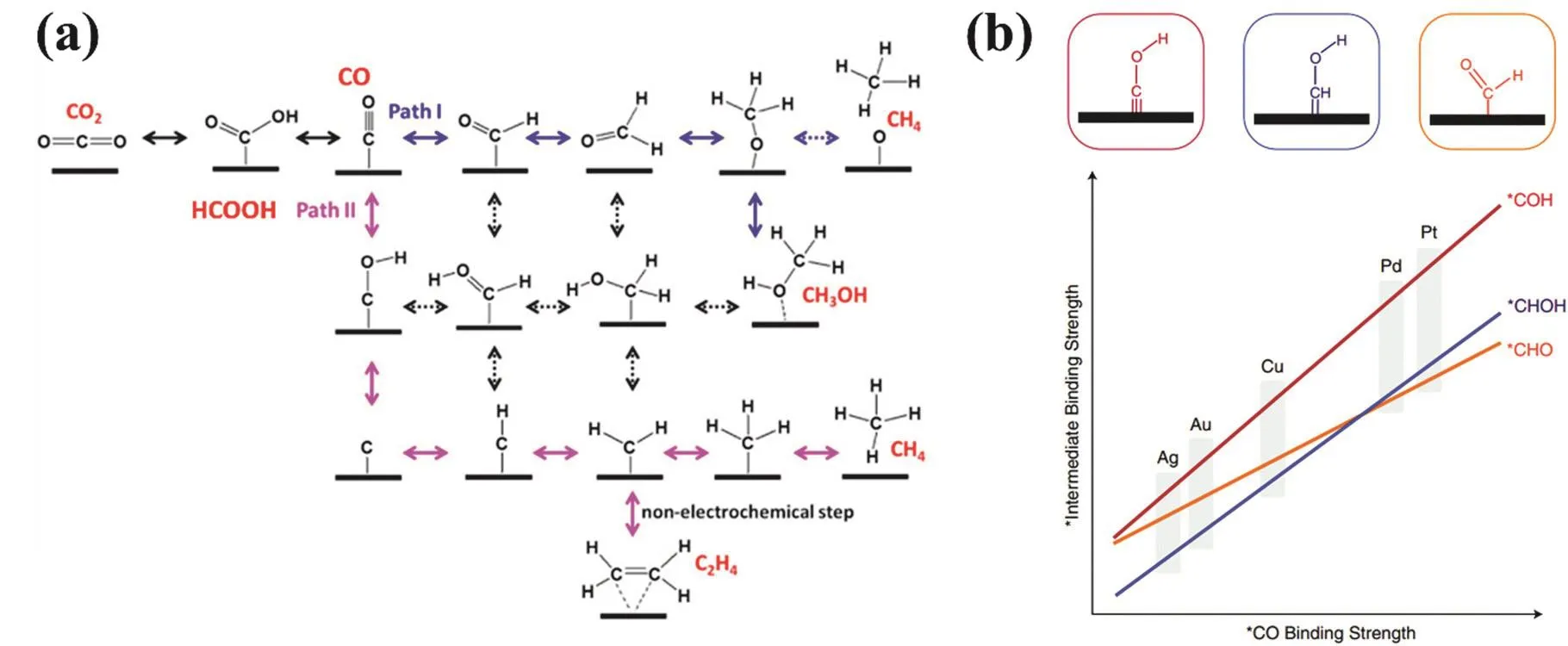
圖1 (a) E-CO2RR不同產物的反應路徑15;(b)催化劑表面*CO與*COH、*CHOH和*CHO吸附能的函數關系的計算結果31Fig. 1 (a) E-CO2RR pathways to different products 15; (b) calculation results of the functional relationship between *CO and *COH, *CHOH and *CHO adsorption energies on catalyst surface 31.
然而,在傳統涂敷催化電極制備過程中,需要將催化劑與粘結劑(如Nafion、聚偏氟乙烯、聚四氟乙烯等)混合均勻后涂覆在集流體上組成功能電極。粘結劑的使用不僅會造成催化劑團聚,還會阻礙電解液與活性位點的接觸,降低活性位利用率和反應效率(圖2a);此外,苛刻的電解液環境或持續運行會致使粘結劑分解和催化劑脫落,導致電極穩定性變差,嚴重制約其實際應用34-36。因此,如何提高電極的活性位利用率和穩定性對于高效催化E-CO2RR至關重要。將催化劑原位有序化(如納米薄膜、納米線、納米柱、納米片、多級結構陣列等)組裝于集流體上得到的一體化電極可避免粘結劑的使用,在E-CO2RR中表現出諸多優勢(以納米柱為例,圖2b):(1)一體化電極可以有效調控三相界面處的微觀反應環境 (如pH、反應物及反應中間體的濃度等),有望打破線性關聯約束、實現多種中間產物的最優吸附37;(2)催化劑原位生長在集流體上可以加速電子在活性位和集流體之間轉移;(3)獨立分散的結構單元可以提供更大的電化學比表面積,暴露更多的活性位點,確保電解液與活性位點的充分接觸;(4)結構單元之間的空間也為電解液的傳輸提供了有效途徑;(5)一體化結構可以增強活性材料與集流體之間的結合強度,提高電極循環穩定性36,38,39。因此,設計一體化電極對E-CO2RR的發展具有重要研究意義。
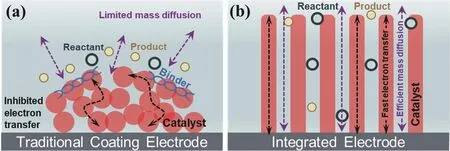
圖2 (a)傳統涂敷電極和(b)一體化電極示意圖Fig. 2 Schematic illustration of (a) traditional coating electrode and (b) integrated electrode.
近年來,研究者們在不同基底(泡沫鎳、泡沫銅、導電玻璃、鎳片、鈦片、碳布、碳紙、碳纖維等)上通過水/溶劑熱法40,41、電沉積法42,43、氣相沉積法44,45和模板法46,47等方法實現了催化劑的有序生長34,35(圖3),并將其直接用于E-CO2RR的工作電極。水/溶劑熱法是制備金屬氧化物和氫氧化物/氫氧化物等無機催化材料的有效方法。在水/溶劑熱反應中,基底上的表面缺陷和羥基是活性物質成核和生長的中心,從而使得納米結構在基底上均勻沉積34,36。Guan等48采用水熱法在泡沫Cu上生長了Bi摻雜的SnO納米片(Bi-SnO/Cu foam),并將其直接作為E-CO2RR合成甲酸(HCOOH)的電極。在-1.7 V vs. Ag/AgCl下,其HCOOH的法拉第效率(Faradaic efficiency,FE)為93%,電流密度為-12 mA·cm-2,能夠穩定運行至少30 h。實驗結果表明Bi的摻雜能夠穩定電催化劑表面二價錫(Sn2+)的存在,使其在電化學還原過程中不易被還原為金屬Sn (Sn0)。此外,結合理論計算發現,Bi的摻雜和電子從催化劑轉移到Cu泡沫基底可以增強*OCHO中間體的吸附,從而有利于HCOOH的生成。電沉積法是另一種制備一體化電極的常用方法。電沉積過程一般在選定導電基底作為工作電極的三電極電化學池中進行,催化材料在外加電場下可以均勻快速地沉積。Jiao等49通過電沉積法合成了具有高活性的Zn枝晶催化電極,其CO轉化電流密度為4-14 mA·cm-2,FE為80%,遠高于體塊的Zn催化劑(0.3-2.8 mA·cm-2,20%)。這表明一體化電極的設計可以顯著提升E-CO2RR性能。氣相沉積法已被廣泛應用于合成一體化電極。其又可分為物理氣相沉積法和化學氣相沉積法,二者的區別在于是否發生化學反應。Kim等50采用微波輔助化學氣相沉積技術制備了含N的金剛石薄膜電極,通過改變生長條件控制sp2-C (石墨)和sp3-C(金剛石)的比例,實現對E-CO2RR活性和選擇性的調控。
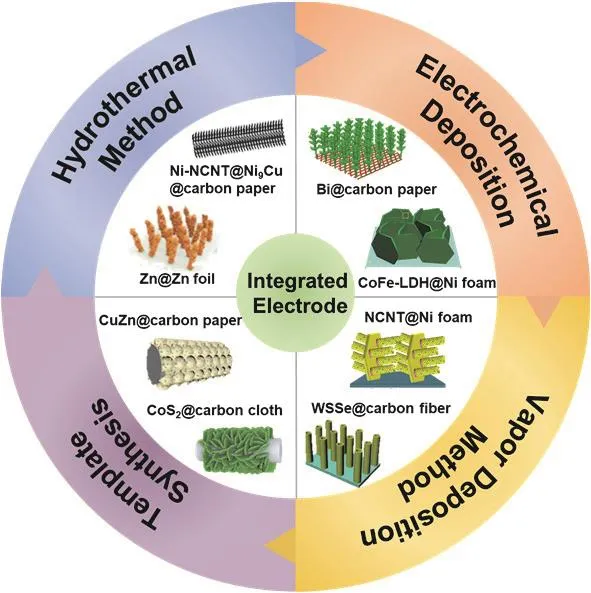
圖3 一體化電極的不同構筑方法40-47Fig. 3 Different construction methods of integrated electrode 40-47.
隨著E-CO2RR相關的基礎研究和應用研究不斷展開,如何從催化劑設計拓展到電極構筑,以及簡化電極構筑方法受到研究者的廣泛關注。但是,目前關于E-CO2RR的綜述多集中在催化劑本征活性調控方面,從電極角度出發的探討和綜述鮮有報道。基于此,本文針對近年來E-CO2RR領域中一體化電極(包括一體化金屬基、一體化金屬化合物基和一體化金屬單原子基E-CO2RR電極)的應用進行了綜述。詳細介紹了不同一體化電極的設計思路,以及結構和表界面調控對E-CO2RR性能影響規律,重點探討了一體化電極對E-CO2RR的構效關系以及性能提升機制等。最后,在現有研究成果的基礎上,對一體化電極在E-CO2RR領域的挑戰和前景進行了展望。
2 一體化金屬基E-CO2RR電極
金屬是研究最早的E-CO2RR電催化劑之一。1980s到1990s之間,Hori等51-53報道了一系列具有開創性的工作,將金屬直接用作電極在KHCO3水系電解質中催化E-CO2RR得到一氧化碳(CO)、甲烷(CH4)、甲酸鹽(HCOO-)和其他碳氫化合物。根據還原產物的不同,將這些金屬分為四類(圖4a):第一類是Au、Ag、Zn、Pd和Ga等。它們能夠結合CO2·-中間體,催化CO2的C―O鍵斷裂,而且CO作為主要的還原產物,很容易從電極表面脫附。第二類是Sn、Pb、Bi和In等。它們很難吸附CO2·-中間體,解吸的CO2·-趨向于在碳原子上質子化,最終還原產物主要為HCOOH或HCOO-。第三類是Pt、Ti、Ni和Fe等。它們具有較低的析氫反應(HER)過電位和較強的CO吸附能,從而使H2成為主要產物。第四類是Cu。在目前報道的眾多金屬中,Cu是唯一可以E-CO2RR生成C1―C3產物的金屬。吸附在Cu表面的*CO傾向于在高過電位下質子化生成*COH或*CHO中間體,進一步還原生成碳氫化合物或者醇類物質7,54-56。
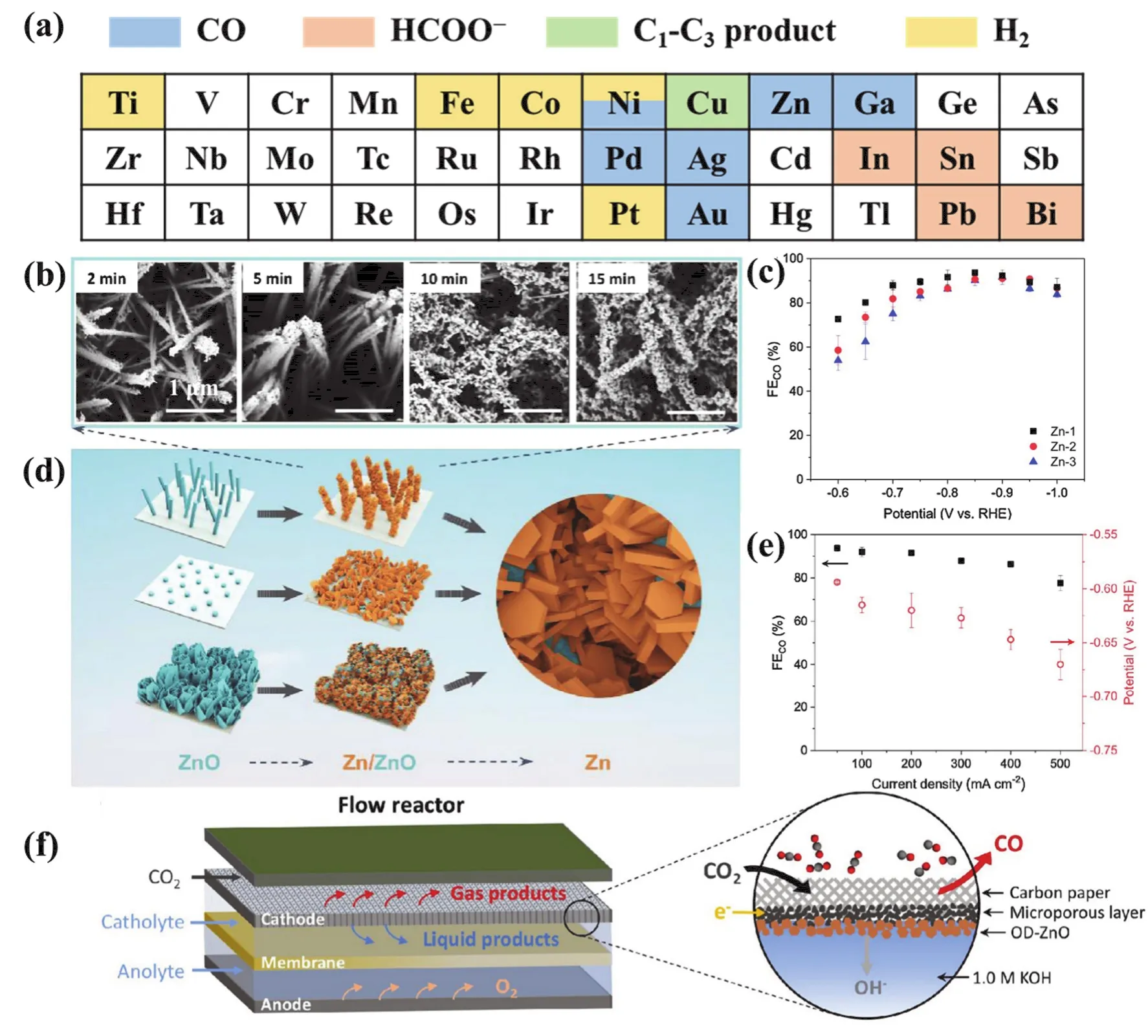
圖4 (a) E-CO2RR金屬分類;(b) CO2飽和的0.1 mol·L-1 KHCO3中-1.6 V vs. RHE時不同還原時間下ZnO-1的SEM圖像;(c)三個ZnO樣品電化學重構過程的示意圖;(d) 0.1 mol·L-1 KHCO3中Zn-1,Zn-2和Zn-3對CO的FE;(e) 1.0 mol·L-1 KOH,液流電池中CO的FE和陰極電壓與電流密度之間的函數關系圖;(f)基于OD-ZnO的氣體擴散電極和液流電池的示意圖40Fig. 4 (a) E-CO2RR metal classification; (b) SEM images of ZnO-1 sample reduced for different time at -1.6 V vs. RHE in CO2-saturated 0.1 mol·L-1 KHCO3; (c) schematic illustration of the electrochemical reconstruction process for three ZnO samples; (d) CO FE of Zn-1, Zn-2 and Zn-3 in 0.1 mol·L-1 KHCO3; (e) FE for CO and cathode potential as a function of current density using 1.0 mol·L-1 KOH in flow cell; (f) illustration of the flow cell and the OD-ZnO based gas diffusion electrode 40.
Pd在極低過電位下可催化E-CO2RR生成HCOO-。Zou等57采用脈沖電沉積的方法將介孔Pd(平均孔徑為~10 nm,壁厚~4 nm)沉積在高度有序的TiO2納米管陣列上,得到了一種新穎的多級結構電極(mPd/TNTAs)。-0.1 V vs. RHE下,mPd/TNTAs催化E-CO2RR至HCOO-的FE為~88% ± 2%,比不含介孔的Pd/TNTAs高2.4倍(35% ± 7%),這得益于mPd/TNTAs中泡沫狀Pd的介孔結構促進了活性位暴露和電解液傳輸。此外,作者通過調控納米管陣列的結構,實現了對納米管內外表面CO2濃度的調節,即一體化電極可以有效調控三相界面處的微觀反應環境(反應物及反應中間體的濃度),實現多種中間產物的最優吸附,進而強化了mPd/TNTAs的E-CO2RR至HCOO-電催化活性,該工作所提出的“結構影響傳質”思想對于結構化電催化劑的設計具有重要借鑒意義。在眾多金屬催化劑中,Zn由于具有儲量豐富、價格低廉等優點,受到廣泛研究。Luo等40通過ZnO的重構衍生制備了不同形貌的Zn電極(圖4b-f)。原位和非原位表征手段表明,和Zn納米顆粒構成的傳統涂敷電極相比,一體化Zn電極表現出顯著提高的比表面積和電化學活性比表面積。從而在催化E-CO2RR時,表現出優異的選擇性和穩定性,其產CO的法拉第效率超過了90%,穩定性超過18 h。將其應用于液流電池中,Zn電極的FE為91.6%,在-0.62 V vs. RHE (可逆氫電極,reversible hydrogen electrode)下電流密度可高達200 mA·cm-2。Bi具有無毒且成本較低的優點,同時在電催化過程中可以很好地抑制HER的發生,被認為是一種很有前途的E-CO2RR電極材料42,58-61。Lee等62采用脈沖電沉積法在Cu基底上原位生長Bi納米薄片。與傳統的直流電沉積得到的Bi膜相比,Bi納米薄片具有大量的邊和角活性位點。數值模擬表明,納米結構的邊或角位點形成了很強的局部電場,增強了其在水溶液中ECO2RR活性。Bi納米薄片在低電位-0.4 V vs. RHE下表現出較高的HCOO-選擇性(FE = 79.5%),-0.6 V vs. RHE時其FE接近100%,表明形貌調控是提高E-CO2RR催化效率的有效途徑。此外,一體化結構擴大了三相反應界面并提高了電極表面CO2濃度,實現催化性能的強化。Sun等63首先在Cu箔表面生長Cu(OH)2納米線,之后進行熱解、電化學還原得到了高密度Cu納米線(Cu NW)陣列電極。經過聚四氟乙烯(PTFE)表面改性后,Cu NW陣列電極具有“親氣疏水”的表面特性(CO2氣泡接觸角為48.4°,電解液接觸角為136.6°),C1產物CO和HCOOH的FE增大(分別約為25%和26%),所有液態產物的FE可達67%。此外,PTFE表面改性后大大抑制了HER的發生(FE < 20%),是無PTFE修飾Cu納米線的一半。
迄今為止,大部分關于E-CO2RR的研究都是在CO2飽和的水溶液中進行(圖5a)。但是,室溫條件下,CO2在水中的溶解度較低(34 mmol·L-1),通過水溶液擴散到電極表面的CO2有限,使得電流密度僅為幾十mA·cm-2,遠低于工業應用所需要的基準(> 300 mA·cm-2)64。通過采用有機電解液、改變電解液壓力和溫度可以提高CO2的溶解度和催化效率,但上述策略無疑會增加催化成本和工藝復雜性,不利于工業化發展31。現階段,H型電解池被廣泛應用于E-CO2RR機理的研究,但其電流密度受到水溶液中CO2溶解度較低和擴散速度較慢的限制,很難滿足工業應用的需求65,66。近期研究表明,通過構建帶有氣體擴散電極(gas diffusion electrode,GDE)的液流電池可以顯著促進CO2的擴散傳輸(圖5b),其擴散路徑僅約為50 nm,遠低于普通H型電解池中的擴散路徑(50 μm)67。氣體擴散電極由一側負載催化劑的氣體擴散層組成,在催化過程中,CO2通過氣體擴散層直接擴散到催化劑表面,E-CO2RR發生在氣-液-固三相界面處。液流電池反應槽中陰極的電解液不斷循環,可以加速反應物擴散吸附到催化劑表面和產物從催化劑表面擴散脫除68。此外,利用液流電池反應槽可以在堿性、高pH的電解質中進行E-CO2RR,這也進一步抑制了競爭反應HER的發生67。一體化電極在構筑氣體擴散電極方面具有明顯優勢,Sargent等69采用電化學拋光的Cu片作為陰極催化劑在H型電解池中測試了其E-CO2RR性能,產CH4的FE和電流密度僅為49%和9 mA·cm-2(圖5c-e)。而在液流電池中,將流動池中局部pH值與H型電解池相匹配后,Cu催化劑催化產CH4的FE可維持在48% ± 4 %,但其電流密度提高至120 ± 10 mA·cm-2,遠大于H型電解池中的電流密度,陰極能量效率可達23%。此外,優化后的液流電池系統在250 mA·cm-2的電流密度下穩定運行14 h,同時維持CH4的FE在40%以上。
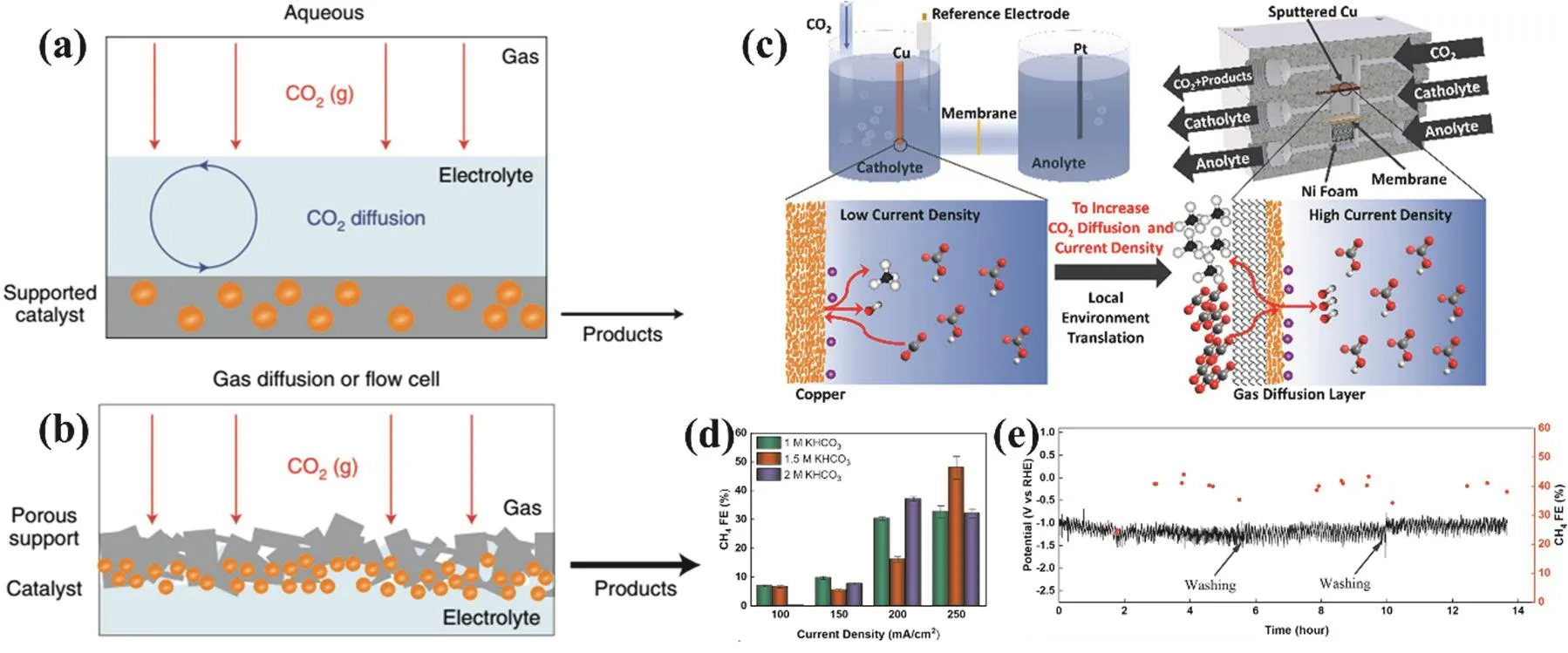
圖5 E-CO2RR溶液的動力學(a) CO2在水溶液中的擴散與溶解;(b) CO2在流動池活性層上直接溶解31;(c) H型電解池和液流電池及其陰極半電池的結構示意圖;(d)液流電池中不同KHCO3下Cu對CH4的FE;(e) Cu 在液流電池 1.5 mol·L-1 KHCO3,250 mA·cm-2下運行 14 h 69Fig. 5 Solution dynamics in E-CO2RR (a) CO2 dissolution and diffusion in an aqueous system; (b) direct dissolution of CO2 at a thin active layer in a flow cell 31; (c) schematic of the H-cell and flow cell configuration and their cathodic half-cell configurations; (d) CH4 FE on Cu with different KHCO3 concentrations in flow cells; (e) 14 h study of Cu by applying 250 mA·cm-2 with a 1.5 mol·L-1 KHCO3 electrolyte in a flow cell 69.
由于E-CO2RR反應路徑和產物繁多,用單一金屬電極調節E-CO2RR的選擇性并抑制副反應HER的發生仍然是一個挑戰。將兩種金屬合金化是提升E-CO2RR性能的有效途徑之一。通過精細調控合金表面的組成和電子結構,能夠改變反應中間體在電極表面的吸附情況22,65,70,71。An等72通過在溫和條件下對Au20Cu80合金進行可控的氧化腐蝕,合成了導電性和柔韌性良好的表面富Au的一體化納米多孔Au3Cu電極。在低電勢(-0.7 V vs.RHE)下,CO的FE高達98.12%,是自制的納米多孔Au電極的1.47倍。Cu的摻雜不僅提高了Au對CO產物的選擇性,還可以降低貴金屬Au的用量。理論計算表明,納米多孔Au3Cu電極對*COOH中間體的吸附能最低,CO解吸所需能量降低。Cu的引入使得Au原子表面帶有更多的負電荷,*CO在距離較遠的納米多孔Au3Cu表面形成穩定的雙齒結構,使得CO更容易從表面解吸,并且避免了CO毒化。實現高電流密度和提高目標產物選擇性是推動ECO2RR工業化應用的重要目標。Sargent等73采用共濺射法合成了一系列不同Ag-Cu比例的Ag/Cu合金作為GDE,電解液為1 mol·L-1KOH的液流電池中,Ag0.14/Cu0.86在-0.67 V vs. RHE時電流密度可達250 mA·cm-2,C2H5OH的FE為41%,陰極能量效率為25%。作為對照,Cu對C2H5OH的FE僅為29%。先前的報道表明Cu表面更容易生成C2H4而不是C2H5OH,通過引入第二種元素Ag,其與碳的結合能力不如Cu,可以降低C2H4中間體在催化劑表面形成的可能性,同時Ag/Cu合金表面更有利于C2H5OH中間體的形成,從而提高C2H5OH的選擇性。
3 一體化金屬化合物基E-CO2RR電極
與金屬相比,金屬氧化物中金屬元素價態易調、富含豐富的界面和缺陷等特征能夠提高ECO2RR電極的本征活性。甚至適當的形貌和氧化程度可能使之前不具備CO2還原催化活性的材料變成一種高活性的E-CO2RR電極74-77。Qiu等78設計合成了泡沫碳支撐的富含氧空位的SnOx納米片電極(VO-SnOx/CF-40),在-1.0 V vs. RHE時,其HCOO-的電流密度為-30 mA·cm-2,FE為86% (圖6a-d)。-1.2 V vs. RHE時,HCOO-的電流密度可達-40 mA·cm-2,其產率高達432.8 μmol·h-1·cm-2。由于SnOx納米片的氧空位和三維的大孔泡沫碳基底的存在,使得其具有更高暴露的活性位點、更快的電子轉移/傳質以及CO2吸附/活化三者之間的協同效應,從而促進了E-CO2RR。Tricoli等79使用熱氣溶膠合成策略在碳纖維紙上直接自組裝得到了3D Bi2O3電極(f-Bi2O3),具有優異的E-CO2RR性能,其起始過電位低至-0.6 V vs. RHE,HCOO-的FE為87%,HCOO-產率為162 μmol·h-1。相比于通過滴涂法得到的Bi2O3傳統涂敷電極(-15.5 mA·mg-1),HCOO-的部分電流密度為-20.9 mA·cm-2(-52.2 mA·mg-1),提高了3倍左右。結果表明,該方法傾向于產生暴露β相的粗糙Bi2O3/Bi電極,其邊緣活性位點的暴露和催化劑獨特的多級結構是獲得高E-CO2RR活性的關鍵因素。此外,原位生長在集流體上可以加速電子在活性位和集流體之間轉移,有利于電流密度的提升。Luo等80在導電碳紙上有序化組裝得到了具有中孔和豐富缺陷的超薄Bi/Bi2O3納米陣列,在-0.87 V vs. RHE下,HCOO-的 電 流 密 度 和 FE 分 別 為 32.4 mA·mg-1·cm-2和90.3%。多級的介孔結構加速了電子轉移,增加了CO2的吸附,縮短了CO2和電解質離子的擴散途徑。此外,Bi/Bi2O3金屬/氧化物結的組成和結構使得局域的電子態有所不同,從而協同促進了CO2的活化和質子化以及還原中間體的穩定。近年來,ECO2RR產CH4僅在較低的電流密度(< 50 mA·cm-2)下才能實現一定的FE(> 50%),遠未達到工業化應用的標準81。采用GDE和液流電池反應槽可以克服CO2在水中溶解度較小和擴散速率較慢的限制,實現電流密度提高至100 mA·cm-2以上的穩定運行。但是,在液流電池系統中高電流密度會使得局部pH值升高,降低水還原的驅動力,并降低催化劑表面對*H的吸附,不利于*CO的質子化,從而抑制了CH4的生成82-86。Sargent等87在Cu表面合成了具有穩定配體的CoO納米簇,在液流電池中,實現了225 mA·cm-2的電流密度和60%的CH4選擇性,并且能夠穩定運行18 h。DFT結果表明,純Cu表面*CO的質子化能為0.43 eV,當局部*H不足時,Cu更傾向于通過二聚生成C2+產物。CoO納米簇的引入提高了局部*H的可利用性,使得Cu表面*CO的質子化更容易發生,提升了CH4的選擇性。
過渡金屬硫化物電極已經在HER88,89、有機小分子氧化90,91、光催化92和超級電容器93等領域展示了廣泛的應用前景,近年來在E-CO2RR中的應用研究與日俱增94-98。Luo等99設計合成了一種泡沫Ni支撐的CuS納米片陣列(CuS @NF)電極,其中CuS納米片的厚度為20-25 nm (圖6e-h)。-1.1 V vs.RHE 時,CuS @NF電極對CH4的FE為73% ± 5%,穩定運行時間可達60 h。除了CuS優異的E-CO2RR本征活性,開放的一體化納米片陣列結構賦予了電極高效的傳質過程和較大的電化學活性面積(Cdl= 39.82 mF·cm-2)。此外,直接生長在泡沫鎳骨架上的CuS納米片具有良好的機械附著力和較高的結構穩定性。He等46提出了一種由直接生長在碳布上的2D CoS2納米片陣列和3D CoS2納米籠組成的多級CoS2納米籠電極。研究表明3D CoS2納米籠/2D CoS2納米片陣列的多級結構不僅可以避免CoS2納米籠的不規則疊加,從而使活性位點充分暴露,還加速了電子在活性位和集流體之間轉移。-0.6 V vs. RHE時,3D CoS2納米籠/2D CoS2納米片陣列產CO的最大FE為85.7%,電流密度為-2.8 mA·cm-2,是粉體CoS2納米籠催化劑的3倍。此外,一體化電極納米片陣列之間的空間也為電解液的傳輸提供了有效途徑,確保了電解液與活性位點的充分接觸。理論計算表明,CoS2的S平面對CO2RR表現出較高的活性,而對HER活性較低;而S邊緣對HER活性高,對CO2RR不利。因此,這種S邊緣較少的多級CoS2納米籠電極大大抑制了HER,從而顯著促進了E-CO2RR的發生。
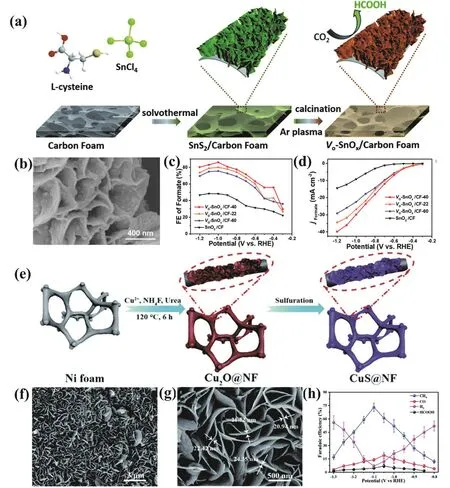
圖6 (a)碳泡沫上富含氧空位的SnOx納米片合成示意圖;(b) VO-SnOx/CF-40的SEM圖像;不同電極對HCOO-的(c) FE和(d)電流密度78;(e) CuS@NF的合成示意圖;CuS@NF的(f)低倍和(g)高倍SEM圖像;(h)不同電壓下CuS@NF產CH4、CO、H2和HCOOH的FE 99Fig. 6 (a) Schematic illustration of the fabrication process of the oxygen vacancy-enriched SnOx nanosheets grown on carbon foam; (b) SEM images of VO-SnOx/CF-40; (c) FE of formate and (d) partial current density of HCOO- for various electrodes 78; (e) illustration of the fabrication process of the CuS@NF sample; (f) and (g) low and high-magnification SEM images of the CuS@NF; (h) FE for CH4, CO, H2 and HCOOH at various applied potentials 99.
除過渡金屬硫化物外,Sun等100在Ti網上合成了FeP納米陣列(FeP NA/TM),直接作為高效的一體化催化電極用于E-CO2RR。FeP NA/TM在-0.2 V vs. RHE時對CH3OH的FE為80.2%,CH3OH和C2H5OH的總FE可達94.3%。在36 h的連續電催化過程中,也表現出非常高的穩定性。理論計算結果表明相鄰兩個Fe原子的協同作用有利于CO2轉化為CH3OH的吸附和還原過程。但是,由于過渡金屬磷化物在水溶液中更傾向于發生HER,因此在E-CO2RR領域報道較少101。
4 一體化金屬單原子基E-CO2RR電極
碳材料具有材料來源豐富、結構易調、表面積大、穩定性高和環境友好等明顯的優勢,其多孔結構也有利于CO2的吸附和電解液的快速滲透4。碳材料負載的單原子催化劑(SACs)由于具有最大的理論原子利用效率、可調的金屬配位結構、獨特的載體效應等,在許多電化學反應中表現出優異的催化性能和選擇性102-107。He等108采用靜電紡絲法設計合成了一種Ni單原子/多孔碳纖維膜電極(NiSA/PCFM),廣泛分布在碳納米纖維中的Ni單原子位點對CO2的活化起著決定性作用,多孔、互相連接的碳納米纖維為CO2擴散和電子傳輸提供了重要通道(圖7a-b)。相比于噴涂粉體法得到的PNiSA/PCFM傳統涂敷電極(FECO= 87%),NiSA/PCFM一體化和多孔結構極大促進了Ni單原子的暴露和利用,從而確保電解液與活性位點的充分接觸,表現出優異的催化活性(FECO= 96%)。此外,一體化結構增強了活性材料與集流體之間的結合強度,提高電極循環穩定性。NiSA/PCFM一體化電極作為GDE應用在液流電池中時,-1.0 V vs.RHE下,其CO的電流密度高達-308.4 mA·cm-2,FE為88%,能夠穩定運行120 h。Wu等109利用氮摻雜的碳和大塊狀金屬Ni固體之間的擴散合成了多級、一體化和原子化的催化電極(H-CPs)。-1.0 V vs. RHE時,H-CPs催化E-CO2RR生成CO的FE高達97%,電流密度為-48.66 mA·cm-2,穩定性超過40 h。在-0.7 - -1.2 V vs. RHE的電勢區間內,FE可維持在90%以上。此外,N-CNTs的垂直排列結構不僅具有優良的導電性,而且其表面的超親水性和超疏氣性,也為離子擴散提供了較大的接觸面積。特別是在高反應速率下,超親水結構有利于通過電解液潤濕催化劑表面進行傳質,而超疏氣表面可以促進氣體產物的脫附。
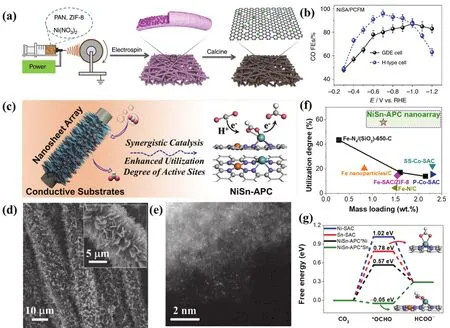
圖7 (a) NiSA/PCFM的合成策略;(b)不同電解池中不同電壓下NiSA/PCFM對CO的FE 108;(c) NiSn-APC示意圖;NiSn-APC的(d) SEM和(e) HAADF-STEM圖像;(f)活性位利用率對比圖和(g) E-CO2RR產HCOO-自由能圖110Fig. 7 (a) Synthesis strategy of NiSA/PCFM; (b) CO FE of NiSA/PCFM at various cathode potentials in different cells 108;(c) scheme illustration for the integrated NiSn-APC electrode; (d) SEM images and (e) HAADF-STEM image of NiSn-APC; (f) a comparative study on utilization degree of NiSn-APC and various materials; (g) free energy diagrams in E-CO2RR to HCOO- on NiSn-APC and references 110.
雖然SACs在E-CO2RR領域受到廣泛關注,但單一SACs活性位點很難滿足具有多電子/多中間產物E-CO2RR的最優催化33。因此,如何通過對催化劑結構進行精細調控來提高催化效率及活性位利用率是亟需解決的關鍵科學問題。我們110提出了一種負載NiSn原子對催化劑的二維碳基陣列一體化電極(NiSn-APC),在E-CO2RR產HCOO-時表現出協同效應,實現了本征活性以及活性位利用率的顯著提升(圖7c-g)。電化學結果表明,所制備的NiSn-APC對E-CO2RR產HCOO-具有優異的催化性能,其HCOO-的產率和轉換頻率分別達到了36.7 mol·h-1·g-1和4752 h-1,在目前報道的Sn基催化劑中處于優勢地位。相比于滴涂粉體法得到的NiSn-APC傳統涂敷電極(FEHCOO-= 62.4%,電流密度為-28.5 mA·cm-2),NiSn-APC一體化電極的FE和電流密度大幅提升,分別為86.1%和-43.7 mA·cm-2。一體化電極的結構優勢加速了電子在活性位和集流體之間的轉移,也為電解液的傳輸提供了有效途徑,進而提升了目標產物的選擇性和電流密度。此外,其Tafel斜率(120 mV·dec-1)明顯小于滴涂粉體法得到的NiSn-APC傳統涂敷電極(164 mV·dec-1),表明一體化的納米陣列結構具有更快的反應動力學,加速了目標產物的生成。活性位定量研究表明,得益于獨特的多級納米片陣列結構,NiSn-APC表現出了高達57.9%的活性位利用率,顯著高于粉體NiSn-APC材料(32.7%)。密度泛函理論(DFT)計算表明Ni-N4的摻雜有利于Sn-N4位點對于HCOO-路徑中間產物*OCHO的吸附,顯著降低了E-CO2RR產HCOO-的反應能壘。Ni-N4可使鄰近Sn原子上的電子離域,從而表現出更高的活性。上述工作表明,通過對多級二維碳納米片陣列表面催化位點進行精細設計,實現了對材料本征活性的可控調控,不僅強化了電極反應過程,還保證了活性位的超高利用率。
5 總結與展望
綜上所述,一體化電極在提升活性位利用率,調控面處微環境以及強化反應穩定性等方面具有重要的優勢,特別是對面向實際應用的催化反應研究起到了重要推動作用。本文從E-CO2RR基一體化電極的設計方法,最新電極種類進展(金屬、合金、金屬氧化物、金屬硫化物/磷化物、金屬單原子等)以及面向高電流密度的電極應用(氣體擴散電極)等角度,對一體化E-CO2RR催化電極進行了系統綜述(圖8,表1)。除此之外,一體化電極在一系列電化學反應,如氧還原反應(ORR)121-124、析氧反應(OER)125-127、HER118,119,128,129、有機小分子氧化90,130,131等中均表現出了優異的催化性能和潛在的實際應用價值。
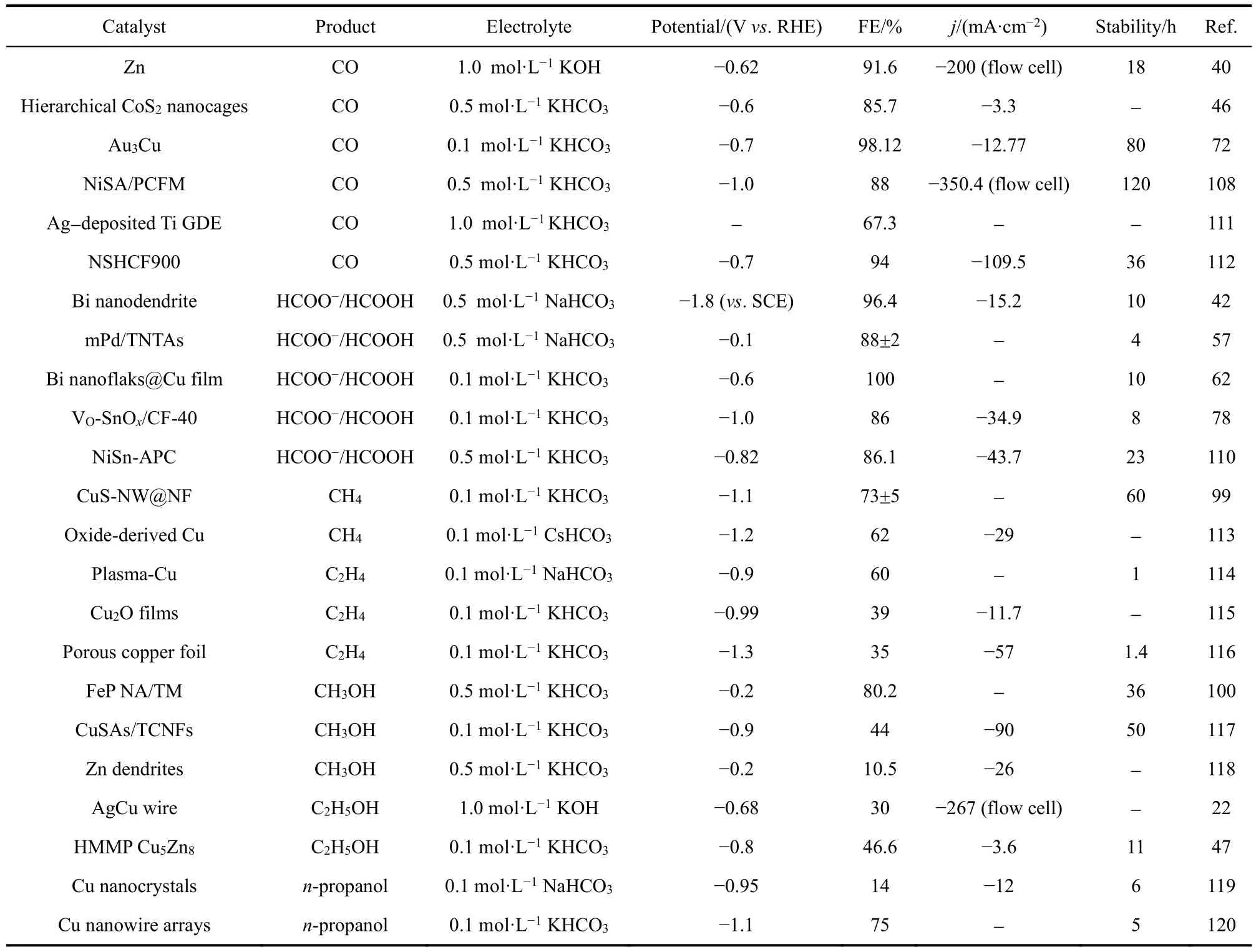
表1 一體化電極E-CO2RR性能匯總Table 1 Summary of integrated electrode for E-CO2RR.
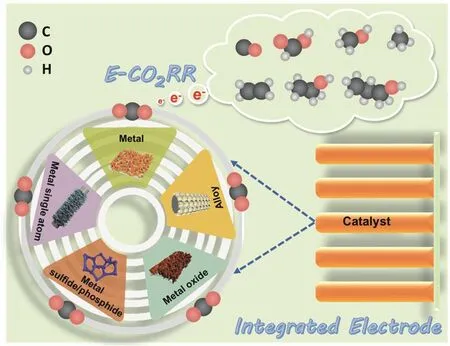
圖8 不同種類的一體化電極用于E-CO2RR 40,47,78,99,110Fig. 8 Different types of integrated electrode for E-CO2RR 40,47,78,99,110.
盡管一體化電極在E-CO2RR方面具有重要潛力,但在基礎研究和應用基礎研究方面仍面臨著機遇與挑戰。
5.1 高效一體化電極合理設計與制備
通過對催化劑的形貌和表界面結構進行精細調控,可以影響催化劑表面CO2濃度、催化劑-電解液界面的pH、反應物和產物傳質過程以及催化劑表面親疏水性等,進而有望改善催化電極ECO2RR的活性、選擇性和穩定性等。目前廣泛使用的制備方法主要包括水/溶劑熱法、電化學沉積法、氣相沉積法和模板法等。上述方法在電極有序構筑、活性位利用、制備成本等方面還需進一步優化。一體化電極如要實現商業化的應用,必然要開發簡便、高效、低成本的合成策略。在各種合成策略中,如何對催化電極的形貌、組成和結構進行更加精細地調控還需要進一步探索。如何在制備過程對所構筑電極的親疏水性進行可控調控,進而研究其與催化性能之間的構效關系仍需進一步探索;如何在生長過程中,加強活性材料與導電基底之間的相互作用,以及研究其對電子傳遞和穩定性的影響機制還需進一步加強;如何通過控制反應條件,實現高負載量一體化電極的制備,以滿足實際應用需求,還面臨巨大挑戰。
5.2 一體化電極界面揭示與反應強化
在實際的E-CO2RR過程中催化電極通常會發生表面重構、形貌轉變、活性位點的演變/湮滅等33,如何采用原位技術(原位X射線衍射(in situXRD)、原位紅外(in situIR)、原位拉曼(in situRaman)和原位X射線吸收光譜(in situXAS)等)132,確定催化活性位點以及對催化電極的動態演變過程和中間產物進行實時監測,深入剖析電極表界面微觀結構變化對催化性能的影響是十分必要的。此外,電解液中的碳酸氫根離子是否參與E-CO2RR,陽離子對E-CO2RR的影響機理以及副反應HER與ECO2RR之間的關系尚不明確。目前,E-CO2RR的理論計算主要依靠熱力學數據,如起始電位、中間體吸附、吉布斯自由能等。現有的實驗結果表明,電極的形貌、局部pH值以及電極-電解液界面的離子種類/濃度對中間體和產物的分布均有影響。然而,上述影響因素在理論計算時很大程度上被忽視了。可用于研究不同電勢下反應電流與活性組分的類型/覆蓋度之間關系的動力學模型也需被考慮其中,這將為一體化E-CO2RR電極的精細設計提供更有利的指導33。
5.3 從一體化電極到新型反應器
雖然H型電解池被廣泛應用于E-CO2RR機理的研究,但其電流密度受到水溶液中CO2溶解度較低和擴散速度較慢的限制。GDE輔助的液流電池能夠有效地將CO2輸送到電極表面,從而滿足工業化的電流密度要求(> 300 mA·cm-2)。但實現穩定運行(> 1000 h),還需要解決GDE的水淹和堵塞問題123。近年來膜電極組裝的電解池也越來越多的應用于E-CO2RR研究。與液流電池不同,膜電極組裝的電解池不需要使用液體電解質,陰陽兩極之間聚合物電解質膜用于離子交換。催化劑直接附著在膜的兩側或者附著在氣體擴散層上置于膜的兩側133-136。受金屬-空氣電池的啟發,金屬-CO2電池(如Li-CO2137,138、K-CO2139和Zn-CO2電池140-143等)近年來也引起了研究者們的關注。但現階段其功率密度大小與理論值還存在一定的差距,電池的安全穩定性也有待進一步的提升。此外,在液流電池或膜電極組裝的電解池中,電池的配置、氣體擴散層、膜、電解質、壓力和溫度等諸多參數對電解池的整體性能都有很大的影響64,144,145。目前,和催化劑電極的設計相比,這些因素沒有得到充分的重視。一體化電極的不斷發展將為未來ECO2RR技術的廣泛應用提供更多的可能。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50

- 物理化學學報的其它文章
- 面向電化學儲能與轉化的表界面工程
- 電壓敏感性聚三苯胺修飾隔膜用于鋰硫電池過充保護
- Surface Modification of NiCo2O4 Nanowires using Organic Ligands for Overall Water Splitting
- NH2-MIL-125 (Ti) Derived Flower-Like Fine TiO2 Nanoparticles Implanted in N-doped Porous Carbon as an Anode with High Activity and Long Cycle Life for Lithium-Ion Batteries
- 三維大孔/介孔碳-碳化鈦復合材料用于無枝晶鋰金屬負極
- 鋰離子電池隔膜的功能化改性及表征技術