男性幼兒雙下肢無力1周伴眼瞼下垂3 d
2022-08-23 00:57:10郎長會田茂強束曉梅
中國當代兒科雜志 2022年8期
郎長會 田茂強 束曉梅
(貴州省兒童醫院/遵義醫科大學附屬醫院小兒內科,貴州遵義 563003)
1 病例介紹
(1)病史:1歲7個月男童,因雙下肢無力1周,伴眼瞼下垂3 d入院。1周前因不能行走、不能站立,逐漸加重到不能獨坐。入院前3 d出現眼瞼下垂,無晨輕暮重,無持物不穩,伴陣發性哭鬧、煩躁不安、精神差及思睡。病程中無飲水嗆咳及聲音低微,無聲嘶及吞咽困難,無發熱、氣促及呼吸困難,無嘔吐、抽搐及大小便障礙。門診以“重癥肌無力(全身型)可能性大”收入院,入院當天完善相關檢查并對癥治療,第2天因考慮為免疫性疾病開始予靜脈注射免疫球蛋白(intravenous immunoglobulin,IVIG)治療,入院第3天(病程第10天)出現陣發性面色青紫、呼吸暫停及呼吸節律不規則,血紅蛋白氧飽和度(oxygen saturation of hemoglobin,SO2)下降至80%,考慮合并中樞性呼吸衰竭轉入兒童重癥監護室(pediatric intensive care unit,PICU)治療。
(2)既往史、個人史、家族史:起病前1月余接種水痘及腦膜炎相關疫苗,前1周有呼吸道感染病史。既往生長發育里程碑正常,入院時可講1~2個字,認知能力正常。患兒系第1胎第1產,出生史及家族史無異常。
(3)入院時體格檢查:T 36.8℃,P 130次/min,R 33次/min,SO298%,嗜睡狀,精神差,哭鬧不安,不能獨坐,雙側眼瞼平視時遮擋角膜位于9~3點方向,眼球運動體格檢查不配合,雙側瞳孔直徑3 mm,光反射靈敏,咽反射存在,頸軟,心肺腹體檢未見異常,雙上肢肌力4級,雙下肢肌力3級,肌張力正常,雙側腱反射消失,布氏征、克氏征陰性,共濟運動體格檢查不配合。
入院第3天轉入PICU時體格檢查:T 36.5℃,P 150次/min,R 20次/min,SO294%(氣囊加壓給氧下),鎮靜狀,呼吸節律不規則,時有呼吸暫停,雙側瞳孔正圓等大,直徑約5 mm,瞳孔對光反射消失。雙上肢肌力約3級,雙下肢肌力約2級,雙側腱反射消失,病理征陰性。
(4)實驗室檢查:血常規、電解質+肝腎功能、血糖、C反應蛋白、肌酶、血氨、乳酸、同型半胱氨酸正常。入院當天(病程第7天)肌電圖未見異常,神經傳導速度(nerve conduction velocity,NCV)無明顯異常。入院第2天(病程第8天)腦脊液常規、生化未見異常。2次新斯的明試驗均陰性。頭顱CT、MRI未見異常。
2 診斷思維
病例特點:(1)幼兒期起病,急性進展性病程,起病前有疫苗接種及呼吸道感染病史;(2)肢體無力進行性加重、眼瞼下垂;(3)病程第5天出現腦病表現(嗜睡);(4)病程第10天出現中樞性呼吸衰竭癥狀(呼吸暫停、呼吸不規則、發紺、SO2下降至80%);(5)既往運動、語言發育正常。入院時體格檢查:嗜睡狀,雙上肢肌力4級,雙下肢肌力3級,雙側腱反射消失。輔助檢查:肌電圖正常,NCV未見明顯異常。
首先定位。(1)下運動神經元,包括前角細胞、周圍神經、神經肌肉接頭、肌肉等。①前角細胞:患兒呈馳緩性肢體無力表現,需考慮前角細胞損害,如腸道病毒感染等,但患兒癱瘓呈對稱性,肌電圖未提示神經源性損害,暫時不支持前角細胞病變,但需進一步行脊髓MRI協助排除。②神經肌肉接頭:患兒以雙下肢無力、眼瞼下垂起病,需考慮有無全身型重癥肌無力,但癥狀無波動性,無晨輕暮重現象,2次新斯的明試驗均陰性,不支持。③肌肉:肌酶正常,肌電圖未提示肌源性損害,可排除。④周圍神經:患兒以雙下肢無力、眼瞼下垂起病,四肢肌力下降,腱反射消失,高度懷疑周圍神經性病變,但患兒NCV未提示潛伏期延長、NCV減慢或動作電位波幅下降及F波異常,也可能在病情早期尚未出現異常,故需進一步動態觀察病情,適時復查NCV。(2)上運動神經元,包括脊髓、大腦等。①脊髓錐體束:因患兒有肢體無力、腱反射消失等表現,需考慮脊髓休克期,但患兒無大小便障礙,且患兒年齡偏小,無法配合感覺體格檢查,進一步查頸胸腰段脊髓MRI協助診斷。②腦干:因患兒除肌無力外還伴有無法解釋的嗜睡,當時無明顯呼吸困難、呼吸無力及缺血缺氧等表現,數天后才出現中樞性呼吸衰竭(呼吸暫停、呼吸不規則及瞳孔散大),需考慮是否累及腦干等。
其次定性。神經系統疾病定性包括感染性炎癥、免疫性炎癥、遺傳性、代謝性、腫瘤性、血管性、中毒性及外傷性等。結合本例患兒急性起病,呈進行性加重,考慮感染性或免疫性炎癥,需排除遺傳性及血管性、中毒性、外傷性病因等。但患兒本次病程中無發熱,輔助檢查炎癥指標不高,故不支持感染性炎癥。急性起病,既往運動、語言發育正常,輔助檢查無明顯代謝紊亂,遺傳代謝性疾病可能性小。患兒急性起病,有眼瞼下垂及肢體無力,但患兒頭顱及脊髓MRI均未見異常,不支持腫瘤性病因。血管性、中毒性、外傷性病因均缺乏相應病史,暫不考慮。本例患兒病前有疫苗接種及前驅感染史,需高度懷疑免疫性炎癥所致,需行相關血及腦脊液抗神經節苷脂抗體(anti-gangliosides antibody,AGA)檢查[1],并進一步動態監測NCV及腦脊液相關指標變化。
3 進一步檢查
入院第3天(病程第9天)復查NCV提示雙側脛神經H反射消失,運動、感覺NCV及F波正常。入院1周(病程第13天)復查腦脊液,提示細胞數仍正常,蛋白增高至1 282 mg/L(參考值200~400 mg/L),提示蛋白細胞分離。同時,外送血清及腦脊液相關抗體,包括水通道蛋白4、少突膠質細胞糖蛋白、星形膠質纖維酸性蛋白、AGA檢測(抗GQ1b抗體IgG和IgM、抗GD1b抗體IgG和IgM,以及抗GM1抗體IgG和IgM),結果顯示血清抗GD1b抗體IgG陽性,腦脊液抗GD1b抗體IgG陰性,余均為陰性。頸胸腰段脊髓MRI未見異常。
4 診斷及診斷依據
診斷:吉蘭-巴雷綜合征(Guillain-Barré syndrome,GBS)、Miller-Fisher綜 合 征(Miller-Fisher syndrome,MFS)及Bickerstaff腦干腦炎(Bickerstaff brainstem encephalitis,BBE)重疊綜合征。診斷依據:(1)GBS:患兒有前驅感染,雙下肢無力進行性加重,在2周達高峰;腱反射消失;腦脊液蛋白細胞分離。(2)不完全性MFS:突出表現的眼外肌麻痹,無其他顱神經受累,伴腱反射消失,無明顯共濟失調表現;伴有腦脊液蛋白細胞分離現象,血清抗GD1b抗體IgG陽性,診斷為不完全性MFS。(3)BBE:眼外肌麻痹;原發病不能解釋的腦病樣表現(嗜睡);中樞性呼吸衰竭(呼吸節律異常)。
5 臨床經過
經IVIG[400 mg/(kg·d)×5 d]、甲潑尼龍沖擊[20 mg/(kg·d)×3 d]、5次血漿置換、呼吸機輔助通氣及對癥支持治療,患兒病情逐漸好轉(眼瞼下垂及嗜睡好轉),精神可。出院時體格檢查:神志清楚,呼吸規則,雙上肢肌力3級,雙下肢近端肌力2級,遠端肌力3級。出院后康復治療半年,患兒可獨立行走、無眼瞼下垂及眼球活動障礙。末次隨訪時(出院后1年),患兒運動基本恢復,行走有力,可跑跳,可上樓。體格檢查示眼球活動正常,無眼瞼下垂,四肢肌力及肌張力正常,腱反射仍未引出。復查NCV示H反射恢復正常。
6 討論
GBS、MFS及BBE重疊綜合征可同時出現周圍神經及腦干損害,臨床異質性強。GBS是由感染后引起的免疫性多發性神經病變,其臨床特征為急性起病、快速進行性對稱肌無力和腱反射減退或消失[2]。眼外肌麻痹、共濟失調和胰反射消失認為是MFS的典型三聯征[3]。GBS和MFS均為感染后急性單相性免疫性疾病,且易伴腦脊液蛋白細胞分離,但二者又有不同的核心臨床癥狀。單純性MFS,單相性病程,4周內達疾病高峰,有典型三聯征,無肢體無力或中樞神經系統受累,并除外其他疾病,腦脊液蛋白細胞分離現象為支持條件。然而,不是每個患兒都有典型三聯征[4],如果缺乏典型三聯征,伴血清AGA陽性及腦脊液蛋白細胞分離,提示不完全性MFS,表現為:(1)不伴共濟失調稱為急性眼外肌麻痹;(2)不伴眼外肌麻痹稱為急性共濟失調神經病;(3)僅表現眼瞼下垂稱為急性眼瞼下垂;(4)僅表現瞳孔散大稱為急性瞳孔散大[5]。BBE以共濟失調、眼外肌麻痹、腱反射消失及腦病為特征,可累及腦干和周圍神經系統。BBE與MFS有一些共同特征如共濟失調、眼外肌麻痹等。BBE除了上述癥狀外,伴有腦病樣癥狀如嗜睡或意識障礙、出現椎體束征[6-7]。本例患兒有典型GBS的臨床表現,如下肢無力進行性加重逐漸累及上肢、腱反射消失、腦脊液蛋白細胞分離,但臨床上還有不能用GBS解釋的癥狀及體征,如眼外肌麻痹,但缺乏后組顱神經麻痹的表現(無聲音變弱、飲水嗆咳及吞咽困難等),考慮重疊MFS;因患兒無明顯的共濟失調表現,考慮為不完全性MFS。此外,患兒有明顯的腦病樣表現(嗜睡)且不能用其他原因解釋(當時無明顯呼吸困難、呼吸無力及缺血缺氧等表現),考慮重疊BBE。
文獻報道外周血AGA在兒童GBS譜系疾病中多以血清抗GQ1b IgG抗體為主,可表現為GBS、MFS及BBE重疊[8-9],較少有抗GD1b抗體陽性的報道。進一步在PubMed(https://pubmed.ncbi.nlm.nih.gov/)或Gene Medical(https://www.geenmedical.com/)中以“GD1b IgG”“GD1b IgM”“children”為關鍵詞,檢索到5篇文獻,共5例患兒[10-14],以男性為主(4/5),多在學齡期起病(年齡5歲5個月至12歲)。臨床表現有肢體無力(2/5),少數出現言語困難、構音障礙及眼外肌麻痹。腱反射正常或減退。輔助檢查可出現腦脊液蛋白細胞分離,神經電生理檢查可出現NCV受限、軸索變性或正常;AGA檢測均有抗GD1b抗體陽性。所有患兒均接受糖皮質激素或IVIG治療(表1)。由此可見,在抗GD1b抗體相關疾病的臨床表現和神經電生理表現存在極大異質性。而本例患兒僅抗GD1b IgG抗體陽性,且臨床表現較文獻中患兒表型更重。也有文獻報道單抗體陽性患者較多抗體陽性患者的臨床表現更重[15]。Kaida等[15]分析同時存在2種或2種以上AGA在細胞膜上形成神經節苷脂復合物,這種神經節苷脂復合物對GD1b抗原的結合活性弱于只有1種抗GD1b抗體的結合活性。這可能是該患兒癥狀比其他患兒更嚴重的主要原因。因此,只有1種抗GD1b抗體在預測疾病嚴重程度方面具有重要意義。
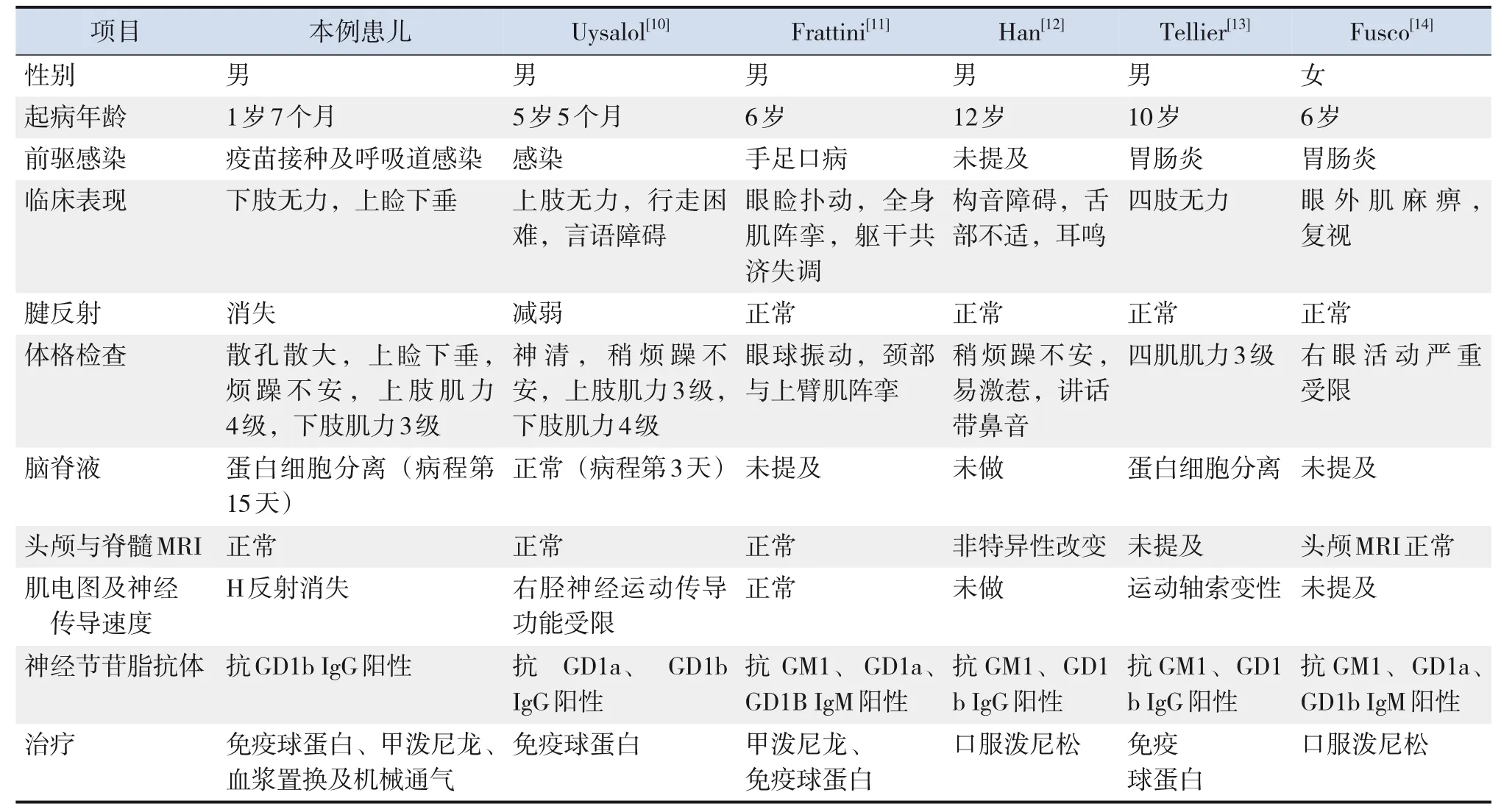
表1 兒童抗GD1b抗體陽性患兒的臨床表型
Kaida等[15]報道成人抗GD1b抗體陽性患者多數以共濟失調和上瞼下垂起病。本例患兒有上瞼下垂,但無明顯共濟失調,分析該患兒的共濟失調可能被嗜睡等癥狀所掩蓋。
本例患兒H反射消失,可能與AGA引起脊髓背根神經元中肌梭或Ⅰa類神經元受損有關,并未引起脫髓鞘或軸突病變,這就可解釋患兒運動/感覺NCV正常,潛伏期或復合肌肉動作電位正常,提示其更容易恢復。Kuwabara等[16]報道約70%的MFS患兒中觀察到H反射缺失。MFS患兒中腓腸神經活檢顯示郎飛結延長,未提示脫髓鞘,可能出現NCV和F波正常,與本例患兒一致。朱瑩等[17]觀察典型GBS及MFS電生理區別,發現典型GBS約有一半表現為運動伴感覺神經傳導異常或單純運動神經傳導異常(27.8%),而MFS組約1/3表現為單純H反射異常,1/3表現為單純感覺傳導異常,還有1/3為正常肌電圖。Rasera等[18]和Dachy等[19]均報道在所有MFS患兒中均有雙側H反射缺失。以上研究結果提示雙側H反射消失在MFS患兒中是一個較靈敏的指標。
GBS、MFS及BBE重疊綜合征的治療包括IVIG或糖皮質激素、血漿置換、康復及對癥治療[4]。關于糖皮質激素治療目前無統一標準,對重癥病例,建議大劑量靜脈滴注甲潑尼龍[20~30 mg/(kg·d)]3~5 d,然后改為口服潑尼松[1 mg/(kg·d)]維持4~6周[20]。本例患兒經IVIG、血漿置換、糖皮質激素及康復等治療后,半年內完全恢復基線水平。
目前認為GBS、MFS及BBE重疊綜合征總體預后較好,約80%的此類患兒在發病6個月后恢復獨立行走能力,3%~10%的患兒死亡,死亡原因最常見是呼吸系統及心血管并發癥[2]。
7 結語
目前臨床醫生對于兒童GBS、MFS及BBE重疊綜合征(尤其在癥狀不典型時)認識不足,且該病有潛在生命危險,若延誤診斷可導致患兒出現呼吸衰竭等嚴重并發癥。當患兒出現四肢無力、上瞼下垂及嗜睡時,應懷疑GBS、MFS及BBE重疊綜合征。應盡快進行NCV檢查,重點關注有無H反射,并完善AGA,早期診斷及治療可改善預后。
利益沖突聲明:所有作者均聲明不存在利益沖突。
