端丙炔酯基環氧乙烷四氫呋喃無規共聚醚的合成與性能研究
2022-09-02 02:05:44劉人天翟進賢楊榮杰宋振坤
火炸藥學報 2022年4期
劉人天,翟進賢,楊榮杰,宋振坤
(北京理工大學 材料學院,北京 100081)
引 言
在固體復合推進劑配方中添加高能填料是提高其能量的重要途徑;以端羥基預聚物為黏合劑、異氰酸酯化合物為固化劑進行交聯反應是制備固體復合推進劑的典型方法。然而,由于異氰酸酯化合物與高能氧化劑二硝酰胺銨(ADN)的化學不相容性,制約了高能復合推進劑的發展。亞銅催化端炔基-疊氮基點擊化學反應(CuAAC)具有反應條件溫和、濕度不敏感、與ADN相容性好等優點,在非異氰酸酯固化交聯制備高能復合推進劑方面獲得廣泛關注。[1-6]
利用疊氮聚醚主鏈上的疊氮甲基基團,Reshmi[7]以端羥基聚疊氮縮水甘油醚(GAP)為黏合劑、兩官能度琥珀酸二丙炔酯(BPS)為固化劑,60℃下固化兩天,通過疊氮與炔基環化反應得到一系列聚三唑交聯GAP熱固性彈性體。Li[8]以端羥基聚3-甲基-3-疊氮甲基氧丁環(PAMMO)為黏合劑、1,4-環己烷二甲酸丙炔酯為固化劑,50℃下放置4天,制得聚三唑交聯PAMMO熱固性彈性體。由于疊氮聚醚與雙官能度炔基固化劑屬無規交聯反應,所得上述彈性體力學性能不佳。Menke、Rahm[9-10]在GAP/BPS體系中加入三羥甲基乙烷三硝酸酯增塑劑,同時添加HMX、ADN等含能固體填料,通過端炔基/疊氮基環化反應得到聚三唑聚疊氮聚醚固體復合推進劑,但力學性能仍不理想。為改善GAP/BPS體系力學性能,Kristensen[11]借助GAP預聚物中同時含羥基、疊氮基的特點,采用雙炔基固化劑和異氰酸酯固化劑,使炔基與疊氮基反應、異氰酸酯與端羥基進行反應,得到含奧克托今固體填料的無煙固體復合推進劑,該體系延伸率有所提高,但玻璃化溫度較高。
疊氮聚醚與炔基固化劑無規交聯難以滿足固體復合推進劑力學性能要求,通過聚三唑末端交聯、制備網絡結構規整的彈性體是改善其力學性能的主要途徑。曲正陽等[12-13]分別以端羥基聚乙二醇400、端羥基聚環氧乙烷-四氫呋喃共聚醚(PET)為預聚物前體,四氫呋喃為溶劑,同時添加丙炔溴、叔丁醇鉀,制備得到了端炔基聚乙二醇(PTPEG)和端炔基聚環氧乙烷-四氫呋喃共聚醚(PTPET);分別將PTPEG、PTPET與疊氮固化劑混合,亞銅催化劑存在下制備得到末端交聯、力學性能良好的聚三唑聚醚彈性體。翟進賢等[14]則將PET預聚物進行端基疊氮化反應,制備得到端疊氮基聚環氧乙烷-四氫呋喃共聚醚(ATPET),隨后將ATPET與三炔丙基胺混合,在亞銅催化劑作用下同樣制備得到末端交聯、力學性能良好的聚三唑交聯聚醚彈性體。由于亞銅化合物易發生氧化或歧化反應而失去催化效果,上述末端交聯反應在實際應用中具有一定的局限性。
提高炔基或疊氮基反應活性,是降低亞銅催化劑依賴、實現炔基與疊氮基非催化反應制備聚三唑末端交聯彈性體的又一前進方向。Pascoal等[15]研究表明,若在炔基官能團附近引入拉電子基團可提高其與疊氮基的反應速率。Patrick等[16]通過競爭反應研究了拉電子效應對炔基化合物反應活性的影響,并且借助密度泛函理論研究了環境化學對炔基/疊氮基環加成反應影響的定量規律。利用羰基拉電子效應,Earla[17]的研究表明炔基單酯、雙酯化合物在室溫下即可與疊氮基團反應生成三唑化合物。Higa[18]的研究表明,磺氧基取代的炔烴化合物與疊氮基在室溫下也可快速反應生成三唑化合物。Qin等[19]將雙(芳酰基乙炔)和二疊氮化物在極性溶劑中混合,非催化條件下制備得到重均相對分子質量26700的聚芳酰基三唑化合物。可以看出,對炔基基團進行化學環境修飾是提高其與疊氮基化學反應效率的一條重要出路,而將該方法應用于聚合物末端改性、以及非催化條件下制備聚三唑末端交聯彈性體的研究還未見報道。
為克服因亞銅催化劑不穩定、易失活而影響疊氮基/炔基反應徹底性的不足,本研究以數均分子質量4000g/mol的端羥基聚環氧乙烷-四氫呋喃共聚醚為反應底物,通過端基酯化反應制備得到了炔基反應活性較高的端丙炔酯基聚環氧乙烷-四氫呋喃共聚醚預聚物(propiolate-terminated ethylene oxide-tetrahydrofuran copolyether,PTPET),并對其性能進行了表征,以期為非催化條件下制備聚三唑交聯聚醚固體復合推進劑原料提供參考。
1 實 驗
1.1 試劑與儀器
端羥基環氧乙烷-四氫呋喃無規共聚醚 (PET,Mn=4038g/mol,羥值為0.435mmol/g)、疊氮縮水甘油醚齊聚物GAP (Mn=480g/mol,平均官能度為3.82),洛陽黎明化工研究院;丙炔酸,純度90%,上海邁瑞爾化學技術有限公司;二環己基碳二亞胺(DCC),純度99%,阿拉丁試劑(上海)有限公司。
Nicolet 6700紅外測試儀,美國熱電公司;AVANCE DRX-500核磁共振波譜儀,德國布魯克公司;Waters Breeze TM2 HPLC System GPC凝膠滲透色譜測試儀,美國Waters公司;Rheostress 300扭矩流變儀,德國Thermo Haake公司;CMT4104電子拉力試驗機,MTS公司。
1.2 實驗方法
室溫下,向裝有機械攪拌裝置的250mL三口燒瓶中依次加入20g PET、3.09g二環己基碳二亞胺、100mL二氯甲烷,機械攪拌至完全溶解。然后逐滴加入1.17g丙炔酸,室溫下反應8h,混合物由淡黃色變為紅棕色。用飽和NaCl水溶液對反應混合物萃取2次,保留下層紅棕色澄清液,加入無水硫酸鈉除水,將液相分離后減壓蒸餾除去二氯甲烷溶劑,真空干燥,得到紅棕色黏稠狀液體端丙炔酯基聚環氧乙烷-四氫呋喃共聚醚PTPET 16.8g。依據PET添加量,所得PTPET產率為84%。
1.3 彈性體的制備
分別制備固化參數為0.8~1.2(疊氮基與炔基的摩爾比)的PTPET/GAP彈性體。將PTPET黏合劑與GAP疊氮固化劑按不同官能團摩爾比混合均勻,倒置于聚四氟乙烯模具中,真空除去氣泡。然后,置于65℃恒溫箱中固化,至混合基體中的疊氮基/炔基紅外吸收峰完全消失,得到反應完全的聚三唑交聯聚醚彈性體樣品。
1.4 性能測試與表征
使用FTIR紅外測試儀對樣品進行紅外掃描,掃描范圍500~4000cm-1,掃描次數32次;利用核磁共振波譜儀對樣品進行核磁13C譜分析,以TMS為內標,CDCl3為溶劑,使用反轉門控去耦對PET和PTPET進行定量測試;將樣品配成10~15mg/mL的四氫呋喃溶液,靜置過夜,以四氫呋喃為流動相,在凝膠滲透色譜測試儀上進行聚合物相對分子質量測試;利用Rheostress 300扭矩流變儀分析聚合物黏度,剪切速率為1~10s-1,每秒采集一次,采集60s;按照GB9865將樣品制成啞鈴狀,通過電子拉力試驗機在室溫下對其拉伸力學性能進行測試,拉伸速率10mm/min。
2 結果與討論
2.1 酯化原理
利用DCC進行酯化反應的機理如圖1所示。反應初始階段,丙炔酸中的活潑氫向DCC中的氮原子進攻,形成中間體。之后,端羥基聚醚上的醇羥基氧原子進攻中間體上的羰基,形成丙炔酸酯化合物。DCC則生成不溶于二氯甲烷溶劑的二環己基脲沉淀,促使反應徹底進行,得到紅棕色目標產物端丙炔酸酯基環氧乙烷-四氫呋喃無規共聚醚PTPET。
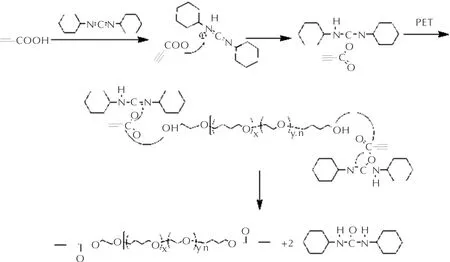
圖1 PTPET酯化反應機理Fig.1 Esterification mechanism of PTPET
2.2 結構表征
2.2.1 FT-IR分析
對原料PET和產物PTPET進行紅外光譜測試,其紅外光譜圖如圖2所示。
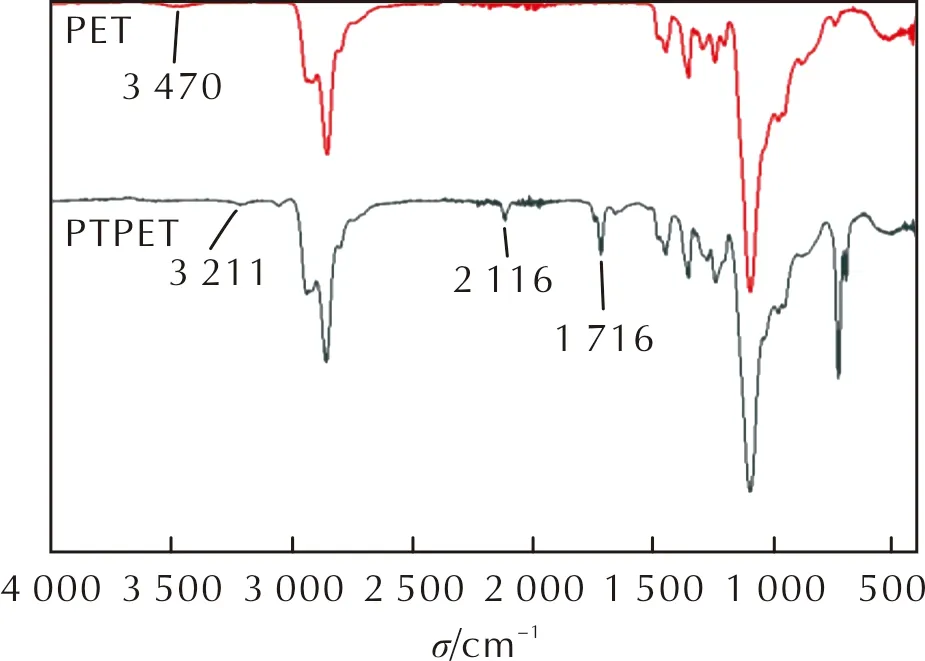
圖2 PET與PTPET的紅外光譜圖Fig.2 FTIR spectra for PET and PTPET
由圖2可以看出,1716cm-1處為丙炔酸酯中C═O伸縮振動吸收峰,2116cm-1處為炔基基團C≡C伸縮振動吸收峰,3211cm-1處為端炔基結構中≡C—H的伸縮振動吸收峰。與PET紅外吸收峰相比,PTPET在3470cm-1處的吸收峰完全消失,表明PET結構中的端羥基已完全酯化,生成了端丙炔酸酯基基團。
2.2.213C NMR 分析
為進一步確認PTPET酯化產物分子結構,對PET和PTPET進行13C核磁測試,所得13C-NMR譜圖如圖3所示。由圖3可知,PET的13C-NMR譜圖中,化學位移70.04~71.12處對應于測點 1和測點 2碳原子, 26.23對應于測點 3碳原子,72.55對應于測點 4碳原子。值得注意的是,與端羥基相連碳原子(測點 5和測點 6)對應于61.72和61.65處共振峰,至77.0處三重共振峰源于溶劑CDCl3。與PET譜圖相比,在PTPET譜圖中,測點 1、測點 2和測點 3碳原子共振峰位置保持不變,仍處于原化學位移處。由于端羥基酯化,測點 4、測點 5和測點 6碳原子共振峰分別移至71.1、74.53處。同時在152.3、152.1處出現測點 7和測點 8碳原子共振峰,76.0、75.1處出現測點 9和測點 10碳原子共振峰。由局部放大圖可以看出,PET中的端羥基在PTPET譜圖中完全消失,表明該酯化反應完全,生成了端丙炔酸酯基環氧乙烷-四氫呋喃無規共聚醚PTPET。
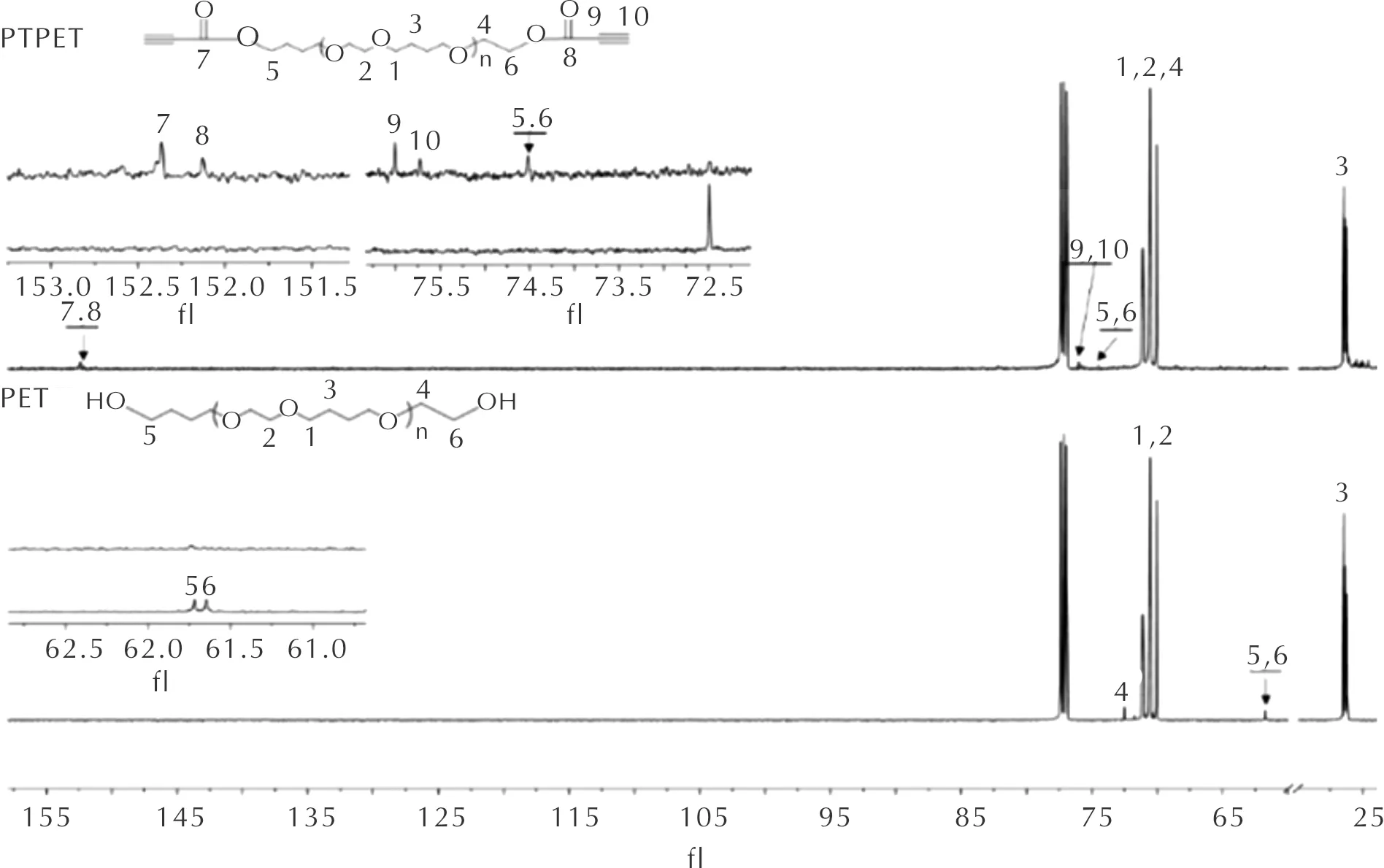
圖3 PET與PTPET核磁13C譜圖Fig.3 13C-NMR spectra for PET and PTPET
2.2.3 GPC分析
為確認PET酯化反應所得PTPET預聚物相對分子質量,對PET和PTPET預聚物進行凝膠滲透色譜測試(GPC),表1是其GPC參數測試結果。

表1 PET和PTPET的GPC結果參數Table 1 GPC parameters for PET and PTPET
由表1可以看出,PET預聚物的數均分子質量為4038g/mol,重均分子質量為6978g/mol,多分散指數為1.73;PTPET預聚物的數均分子質量為4153g/mol,重均分子質量為7177g/mol,多分散指數為1.73,PET與PTPET兩者相對分子質量參數非常接近。
圖4為PET與PTPET預聚物GPC流出曲線。由圖4可以看出,PET與PTPET的GPC流出曲線位置和峰型基本一致。結合表1測試結果可以推斷,PET端羥基與丙炔酸酯化過程中,僅僅是PET預聚物的端羥基發生了酯化,生成了PTPET,而聚合物并未發生斷鏈、擴鏈等副反應。PTPET與PET的端基官能團含量相一致。
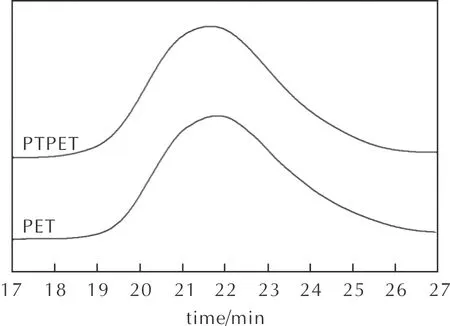
圖4 PET與PTPET的GPC曲線Fig.4 GPC curves for PET and PTPET
2.3 黏度分析
PTPET黏度是其重要參數之一,直接影響其使用性能。為揭示端羥基丙炔酸酯化對共聚醚黏度的影響,利用Haake流變儀對PET和PTPET預聚物黏度進行了測試,圖5為其剪切速率—黏度關系曲線。由圖5可以看出,相同剪切速率下,PTPET預聚物黏度顯著高于PET。鑒于預聚物PTPET與PET兩者的相對分子質量及其分布極為接近,兩者的唯一區別源于其端基官能團,因此PTPET預聚物的高黏度特性應源于其分子鏈的端位基團。對于丙炔酸酯基基團,羰基基團與炔基的三鍵可形成p軌道電子共軛結構,由于羰基結構中氧原子的拉電子效應,使得端炔基結構中的氫原子更加活潑,可與其他丙炔酸酯基上的羰基基團形成氫鍵[20],增加了PTPET分子鏈間的相互作用,PTPET分子鏈段運動阻力增加。因此,PTPET聚合物黏度顯著高于PET體系。
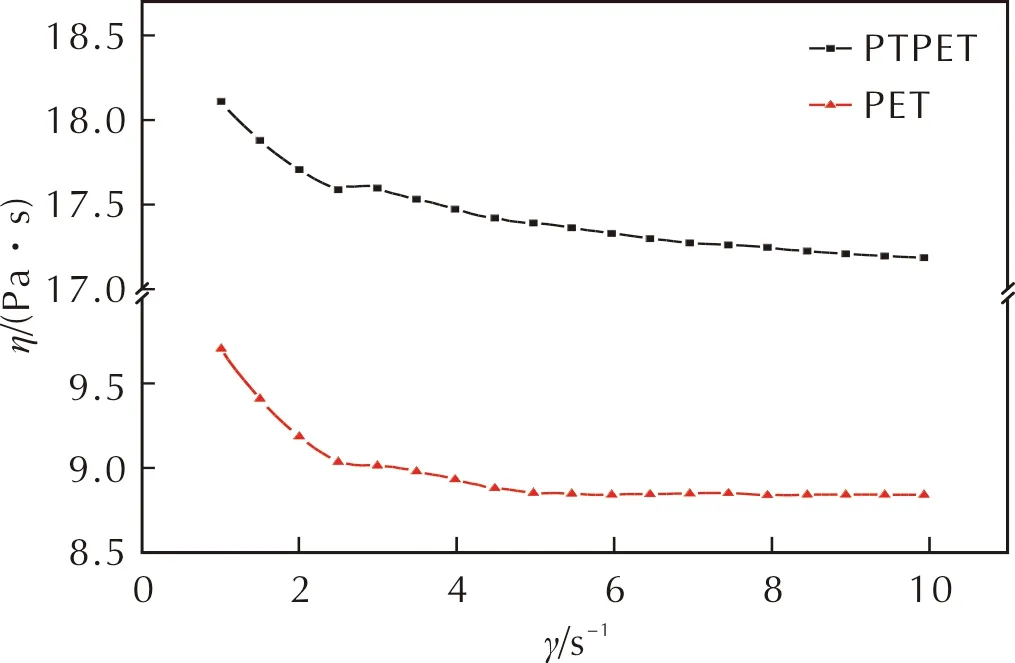
圖5 PET與PTPET剪切速率—黏度關系曲線Fig.5 Shear rate—viscosity curves for PET and PTPET
2.4 端炔基含量的確定
PTPET端炔基含量是確定固化劑GAP添加量、調節固化參數、獲得不同網絡交聯結構彈性體的關鍵參數。目前,有關聚合物端炔基含量的定量測定方法尚未見文獻報道,而通過聚合物前驅體官能團等價換算是確定聚合物端炔基含量的一種簡潔、快速的方法。鑒于PTPET預聚物分子鏈結構中端羥基被完全酯化(見13C-NMR分析),且PET聚合物分子鏈在酯化過程中未發生斷鏈、交聯等副反應(見GPC分析),因此,可以推斷PTPET聚合物中的端炔基摩爾數與PET中端羥基摩爾數相同,依據式(1)可計算獲得PTPET預聚物的端炔基摩爾含量。
CAlkyne=CHydroxyl/(1+CHydroxyl×52)
(1)
式中:CAlkyne為PTPET中炔基的含量,mmol/g;CHydroxyl為PET中羥基的含量,mmol/g;52為丙炔酯基摩爾質量69g/mol與羥基摩爾質量17g/mol的差值,g/mol。
將產物PTPET前驅體PET羥值(0.435mmol/g)代入式(1),計算得PTPET的端炔基值為0.425mmol/g。
2.5 彈性體力學性能分析
依據上述2.4節確定的PTPET炔基值,將其與GAP固化劑按不同比例混合,制備不同R值(GAP中疊氮基與PTPET中炔基比例)的聚三唑交聯聚醚彈性體。表2是該彈性體室溫下力學性能測試結果。
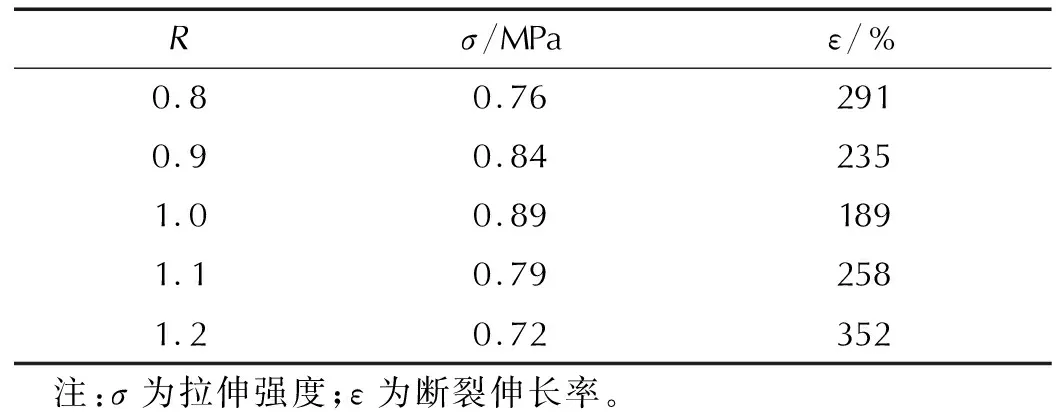
表2 聚三唑交聯彈性體的室溫力學性能Table 2 Mechanical properties of polytriazole elastomers at room temperature
由表2可以看出,在R值為1時,彈性體拉伸強度為0.89MPa,斷裂伸長率為189%,在5組彈性體中具有良好的力學性能。
3 結 論
(1)以PET為原料,通過端羥基酯化反應制備得到了端丙炔酯基環氧乙烷-四氫呋喃無規共聚醚目標化合物PTPET。FTIR、13C-NMR分析表明,PTPET分子結構中與端羥基有關的紅外特征吸收峰和核磁共振峰完全消失,產物丙炔酸酯化反應完全。
(2)PTPET具有與前驅體PET相近的相對分子質量及其分布特性。相近相對分子質量條件下,PTPET較PET具有較高的黏度。
(3)非催化條件下,PTPET與GAP可交聯固化形成聚三唑交聯彈性體,力學性能良好,為制備聚三唑聚醚復合推進劑拓寬了原料基礎。
