水產品中硝基呋喃類藥物代謝殘留的不確定度評定
2022-09-08 01:31:16呂燕
浙江農業科學 2022年9期
呂燕
(寧波市農業科學研究院,浙江 寧波 315040)
硝基呋喃類抗生素(nitrofuran antibiotics) 是人工合成的抗感染類藥物[1-3],主要是指呋喃唑酮(furazolidone)、呋喃西林(nitrofurazone)、呋喃妥因(furantoin)和呋喃它酮(furaltadone),及其對應的代謝產物有呋喃唑酮(AOZ)、呋喃西林(SEM)、呋喃妥因(AHD)和呋喃它酮(AMOZ)。這類抗生素被廣泛用于家禽、家畜、水產、蜂等動物傳染病的預防與治療,但有關研究證明[4-9],硝基呋喃類藥物及其代謝物具有較強的毒副作用,可能誘導有機體基因突變,有致畸和誘發癌癥的可能,從而被人們高度重視。
如今,我國及大多數歐美國家都禁止使用該類藥物,為確保該藥物日常檢測結果的準確,本文利用《化學分析中不確定度的評估指南》[10]和《測量不確定度評定與表示》[11]等相關技術標準的規定,對用農業部783號公告-1-2006[12]的方法進行前處理并分析檢測過程中可能產生的不確定因素,評定最終檢測結果的不確定度[9,13-15],以保證最終結果的準確與可靠。
1 材料與方法
1.1 材料
儀器:液相色譜/串聯質譜儀、組織搗碎機、分析天平、渦旋振蕩器、氮吹儀、容量瓶、離心管、移液槍等。
試劑:甲醇、乙酸乙酯、乙酸銨等。除特殊說明外,其他試劑均為分析純。實驗用水為超純水。
標準品:AOZ、AMOZ,SEM、AHD,純度均大于99%。同位素內標:AOZ-D4、AMOZ-D5,SCA-15N2-13C HCL、AHD-13C3,純度均大于99%。
1.2 方法
1.2.1 標準溶液的配制
混合標準工作溶液的配制:稱取AMOZ、AOZ、AHD和SEM標準品各10 mg,用甲醇溶解得到100 mg·L-1的AMOZ、AOZ、AHD和SEM標準儲備液。吸取1.0 mL AMOZ、AOZ、AHD和SEM標準儲備液用甲醇配制得到1.0 mg·L-1AMOZ、AOZ、AHD和SEM的混合中間標準溶液。再吸取0.1 mL 1.0 mg·L-1AMOZ、AOZ、AHD和SEM的混合中間標準溶液,用水稀釋得到0.01 mg·L-1AMOZ、AOZ、AHD和SEM的混合標準工作溶液。內標標準溶液的配制與混合標準溶液的配制方法一致。
1.2.2 樣品前處理方法
按照農業部783號公告-1-2006《水產品中硝基呋喃類藥物代謝物殘留量的測定液相色譜-串聯質譜法》的樣品處理方式進行提取。
1.2.3 液相色譜-串聯質譜條件
色譜柱為C18,液相色譜流速及梯度洗脫程序見表1。采用正離子掃描方式,多反應監測模式(MRM),質譜定性定量離子對信息見表2。

表1 液相色譜梯度洗脫條件
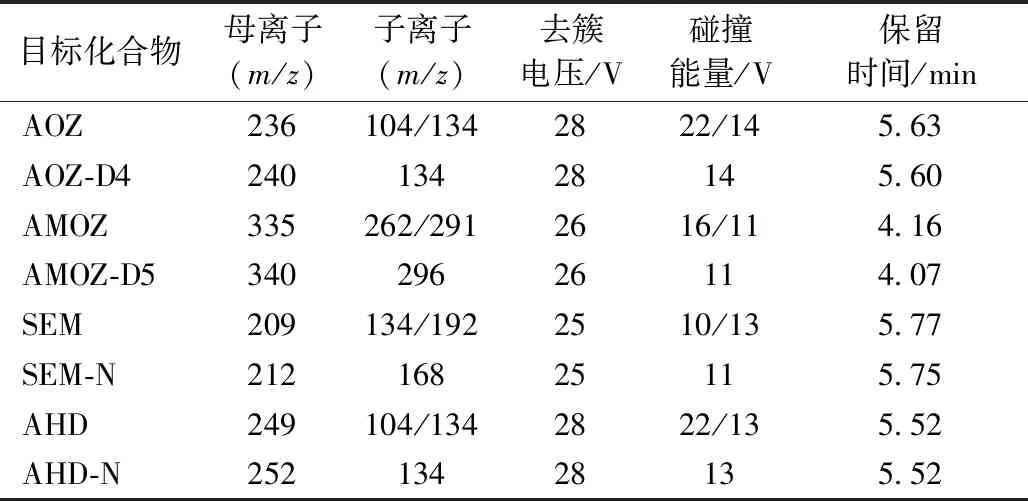
表2 目標化合物液質測定條件
2 結果與分析
2.1 數學模型的建立
根據農業部783號公告-1-2006測定方法及結果計算公式,建立樣品中硝基呋喃類代謝物殘留量測定的數學模型如下:
X=RcV/Rsm。
(1)
式(1)中:X為檢測樣品中硝基呋喃類代謝物的含量,μg·kg-1;R為提取的樣品待測液中的目標物與內標峰面積比;c為混合標準溶液中硝基呋喃類代謝物的質量濃度,ng·mL-1;V為提取的樣品待測液最終的定容體積,mL;Rs為標準液中的目標物與內標峰面積比;m為檢測樣品的質量,g。
以上各種因素都會對測量結果的不確定度產生影響,其最終合成相對標準不確定度計算公式為:
(2)
式(2)中:urel(c)為合成相對標準不確定度;urel(c0)為標準曲線擬合引入的相對標準不確定度;urel(cs)為標準溶液配制的相對標準不確定度;urel(m)為樣品稱量的相對標準不確定度;urel(V)為樣品前處理過程引入的相對標準不確定度;urel(frep)為重復測量的相對標準不確定度;urel(R)為加標回收率的相對標準不確定度。
2.2 不確定度來源分析
從測定過程和數學模型分析,硝基呋喃類殘留測定的不確定度來源如圖1所示。
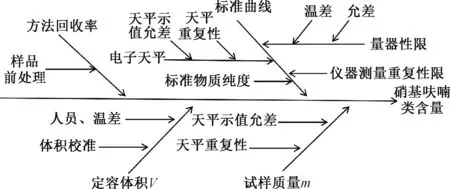
圖1 不確定度來源的因果關系
本次測定的不確定度主要來源于以下幾個方面:測量重復性引起的不確定度;曲線校準引入的相對標準不確定度;樣品前處理過程引入的不確定度;標準物質配制過程中的不確定度;方法回收率引入的不確定度。
2.3 相對標準不確定度分量的評定
2.3.1 標準曲線擬合引入的相對標準不確定度urel(c0)
本實驗對4種硝基呋喃類代謝物,選取與硝基呋喃類代謝物相應的同位素內標濃度比,制備了不同濃度點的標準工作液,每個濃度點校準溶液平行測定3次,以標準溶液濃度Ci為橫坐標,以標準溶液峰面積比Ai為縱坐標,采用最小二乘法擬合,獲得線性回歸方程(Y=a+bX)以及線性相關系數r,如表3所示。
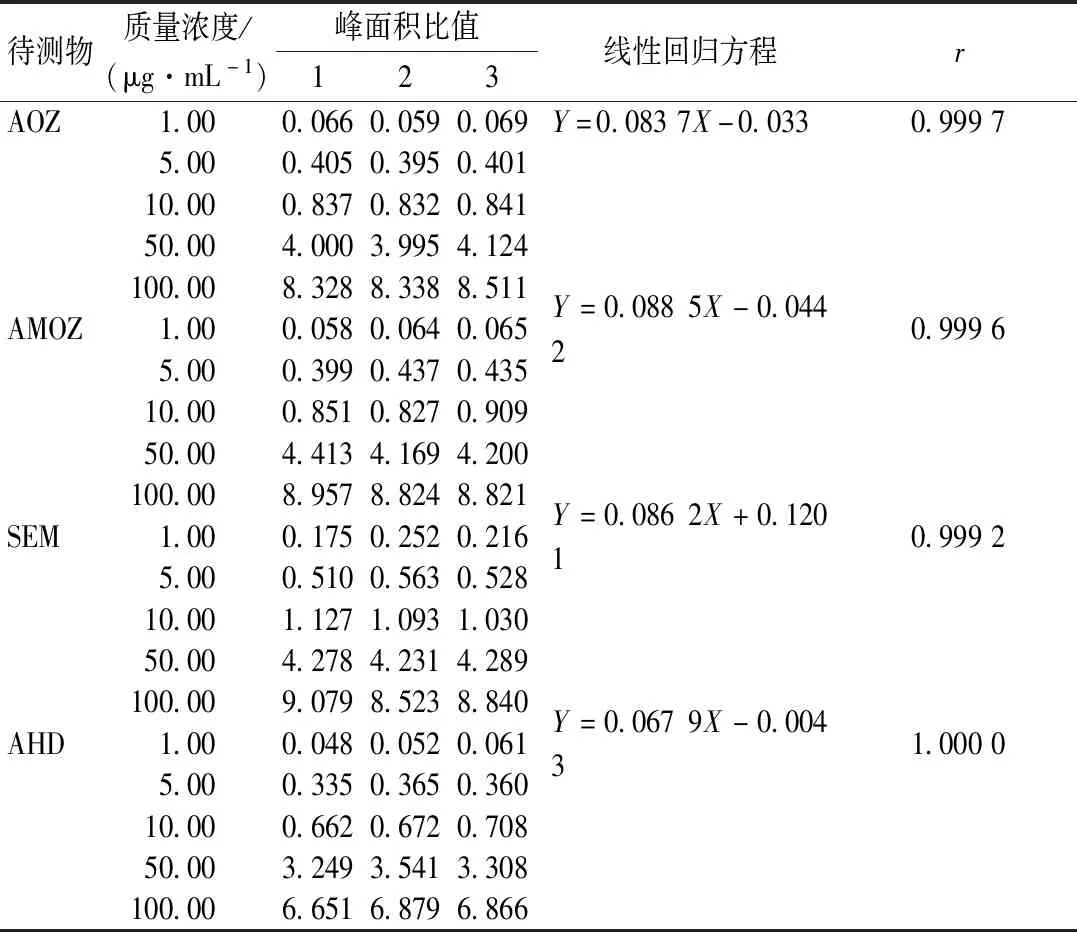
表3 硝基呋喃類代謝物回歸方程測量結果
線性回歸標準曲線擬合引入的不確定度公式如下:
(3)
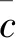
標準曲線擬合引入的相對標準不確定度urel(c0)結果見式(4)。
(4)
相對不確定度結果列于表4。
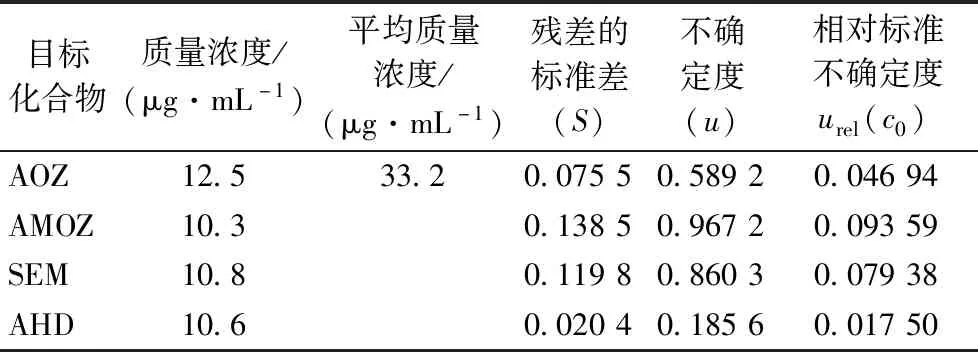
表4 相對不確定度
2.3.2 標準溶液配制過程引入的相對標準不確定度urel(cs)
標準溶液配制過程引入的相對標準不確定度的數學模型為:
(5)
標準物質純度引入的相對標準不確定度見表5。
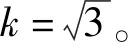
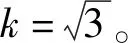
表中:

(6)
根據以上計算的各分量及公式(5)可以計算出標準溶液配制過程中的相對標準不確定度urel(cs):
2.3.3 樣品稱量過程中引入的相對標準不確定度urel(m)
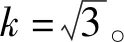
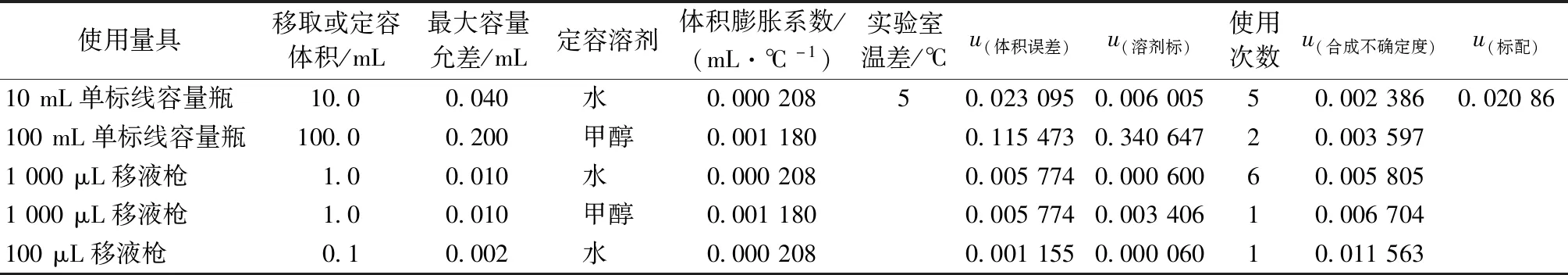
表6 標準曲線配制引入的不確定度
(7)
2.3.4 樣品前處理過程中引入的不確定度urel(V)
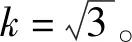
(8)
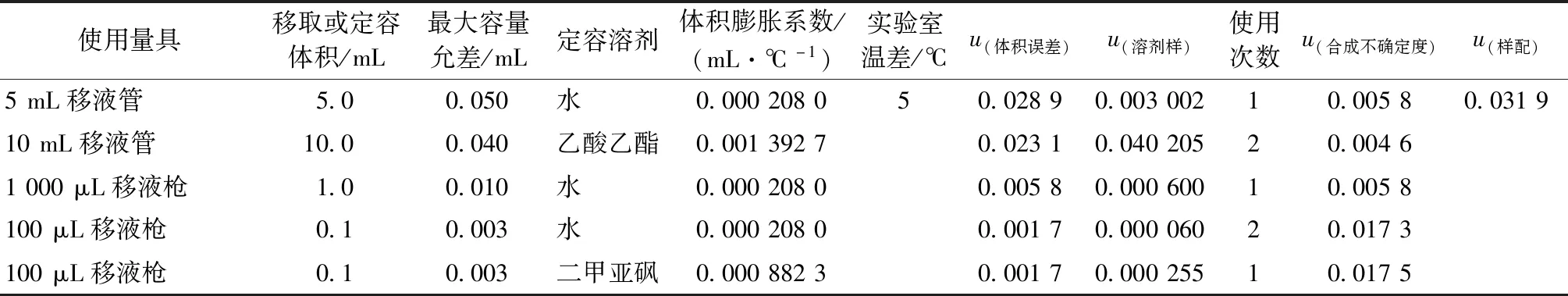
表7 樣品制備過程引入的不確定度
2.3.5 整個實驗重復性引起的相對標準不確定度urel(frep)
整個實驗包括樣品稱量操作的重復性和進樣體積的重復性等。因此,將這些重復性分量合并為總試驗的一個分量,利用方法確認的數值將其量化是合理的。稱量的重復性屬A類標準不確定度,以算數平均值的標準偏差表示。按照1.2節方法進行處理和上機測定,平行測定樣品6次,用貝塞爾公式(9)進行計算,結果見表8。公式如下:
(9)
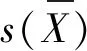
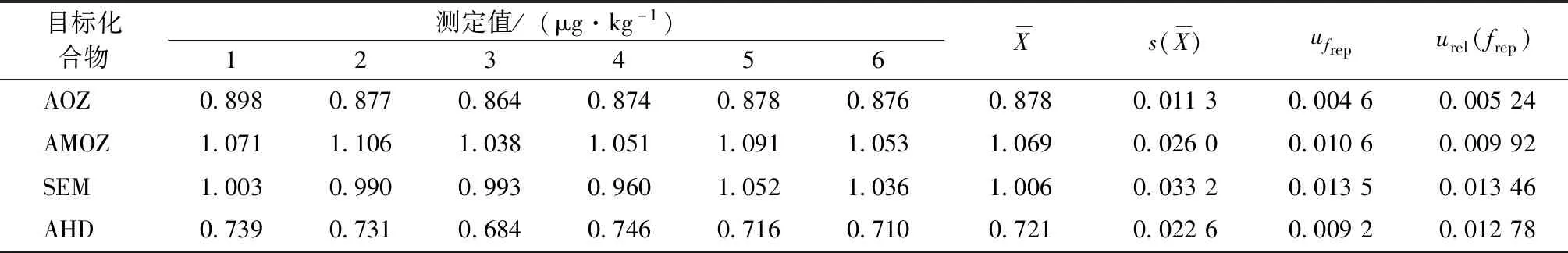
表8 重復測定結果及其相對標準不確定度(n=6)
2.3.6 加標回收率引入的相對標準不確定度urel(R)
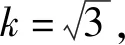
(10)
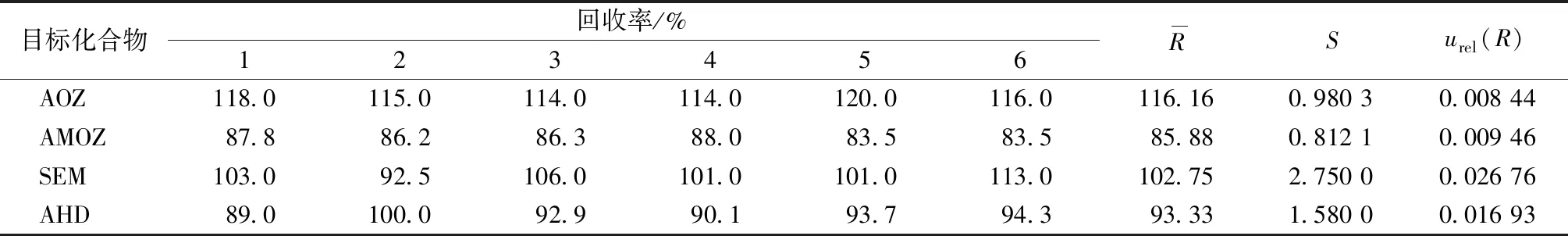
表9 加標回收率結果和加標實驗引入的相對標準不確定度
2.3.7 合成相對標準不確定度urel(c)及不確定度uc(X)
以上各項不確定度分量相對獨立,不考慮分量間的相關性,將上述各值代入公式(2)中,得出合成相對標準不確定度urel(c),再利用公式(11)來計算不確定度uc(X),最終檢測結果取包含因子結果合成不確定度,結果如表10所示。
uc(X)=urel(c)·X。
(11)
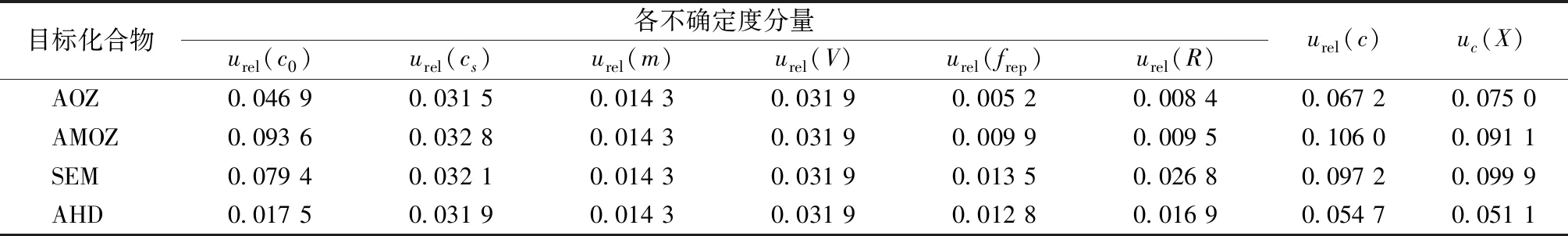
表10 合成相對標準不確定度和不確定度
2.4 擴展不確定度
取包含因子k=2(近似95%置信概率),擴展不確定度U=kUc(X),則結果可報告為:X(AOZ)=1.161 6 μg·kg-1,U=0.150 μg·kg-1,k=2;X(AMOZ)=0.858 8 μg·kg-1,U=0.182 μg·kg-1,k=2;X(SEM)=1.027 5 μg·kg-1,U=0.200 μg·kg-1,k=2;X(AHD)=0.933 3 μg·kg-1,U=0.102 μg·kg-1,k=2。
3 小結與討論
本文按照農業部783號公告-1-2006《水產品中硝基呋喃類藥物代謝物殘留量的測定液相色譜-串聯質譜法》對硝基呋喃類藥物進行檢測,通過建模、評定和計算測量整個實驗過程的各個不確定度分量,得出最終擴展不確定度。結果表明,硝基呋喃類代謝物殘留量測定的不確定度主要來自于標準溶液和內標溶液的配制及使用、標準曲線的擬合、加標回收率,其次為測量重復性和樣品稱樣量。因此,在日常檢測過程中,應盡量選擇高精度器皿,并適當增加測定次數,以最大限度降低整個檢測過程中可能帶來的不確定度影響,提高檢測結果的準確性,為正確評價和使用檢測數據提供依據。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
中學生數理化·七年級數學人教版(2020年12期)2021-01-18 06:57:46
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
海峽科技與產業(2016年3期)2016-05-17 04:32:12
