人發(fā)角蛋白透析純化研究
2022-09-08 06:39:30郭克兵田湞楨付艷紅盧伊婷季金茍郝石磊
大眾科技 2022年8期
關(guān)鍵詞:結(jié)構(gòu)
郭克兵 田湞楨 付艷紅 盧伊婷 季金茍 郝石磊
人發(fā)角蛋白透析純化研究
郭克兵1田湞楨1付艷紅1盧伊婷1季金茍1郝石磊2
(1.重慶大學(xué)化學(xué)化工學(xué)院,重慶 401331;2.重慶大學(xué)生物工程學(xué)院,重慶 401331)
為了了解透析純化過程中人發(fā)角蛋白二級(jí)結(jié)構(gòu)的變化情況,用接觸角測(cè)定儀、圓二色譜儀(CD)和X-射線衍射儀(XRD)等對(duì)高溫氨解提取的人發(fā)角蛋白進(jìn)行了分析。結(jié)果表明,透析0 h~36 h,角蛋白親水性減小,二硫鍵含量增加,α-螺旋含量顯著增高,無規(guī)則卷曲含量占比明顯減少;透析36 h~72 h,親水性依舊減少,二硫鍵含量基本不變,無規(guī)則卷曲占比顯著增大。透析過程對(duì)α-螺旋和β-折疊晶區(qū)基本沒有影響。
人發(fā)角蛋白;透析;純化;時(shí)間;結(jié)構(gòu)
引言
人發(fā)角蛋白因其生物相容性、良好的組織黏附性被廣泛用于制作水凝膠、薄膜、支架等生物材料[1-3]。人發(fā)角蛋白通常從人發(fā)中提取而來[4,5],因此人發(fā)角蛋白的提取純化過程受到越來越多的關(guān)注。
人發(fā)角蛋白是以α-螺旋為主要結(jié)構(gòu)的α-角蛋白,屬于不溶性大分子,因此難以提取[6-8]。研究者們開發(fā)了包括機(jī)械法、還原法、氧化法等方法的提取方法[4,5],但是提取過程往往包含透析純化步驟。純化是角蛋白應(yīng)用前的必經(jīng)步驟,通常透析純化大多固定在一定時(shí)間,對(duì)其研究較小。
透析技術(shù)是最簡(jiǎn)便、最常用的分離純化技術(shù)之一,可以去除鹽、少量有機(jī)溶劑和小分子雜質(zhì),并使樣品濃縮和改變微量樣品的緩沖系統(tǒng)等。用透析袋透析簡(jiǎn)單方便、廉價(jià)、可循環(huán)利用,即將生物大分子樣品溶液置入袋內(nèi),浸入水或緩沖液中,樣品溶液中的大分子量的生物大分子被截留在袋內(nèi),而鹽和小分子等物質(zhì)不斷擴(kuò)散透析到袋外,直到袋內(nèi)、外兩邊的濃度達(dá)到平衡。一般認(rèn)為,角蛋白在透析過程中其二級(jí)結(jié)構(gòu)會(huì)發(fā)生一定的變化,但是對(duì)變化過程未有深入的了解,現(xiàn)代研究發(fā)生,角蛋白的二級(jí)結(jié)構(gòu)對(duì)其生物應(yīng)用具有重要影響,因此,本文采用高溫水氨解自制的人發(fā)角蛋白,對(duì)其透析純化過程進(jìn)行相關(guān)研究。
1 材料與方法
1.1 試劑與儀器
人發(fā)(搜集于理發(fā)店);聚氧乙烯辛基苯酚醚-10(OP-10)(成都科龍化工試劑廠);氫氧化鈉、23%氨水、丙烯酰胺、三羥甲基氨基甲烷(Tris)(重慶川東化工有限公司);碳酸氫鈉(成都市科隆化學(xué)品有限公司);十二烷基硫酸鈉(SDS)(美國Sigma公司);考馬斯亮藍(lán)R250、過硫酸銨,(上海生工生物工程股份有限公司);醋酸(國藥集團(tuán)化學(xué)試劑有限公司)。以上所有試劑均為分析純。二硫代雙硝基苯甲酸(DTNB),生物試劑(上海阿拉丁生化科技股份有限公司)。蛋白上樣緩沖液、彩色預(yù)染marker(11-180 kDa),生物試劑(上海源葉生物科技有限公司)。實(shí)驗(yàn)用水為實(shí)驗(yàn)室自制去離子水。
GFK-10-200型水熱合成反應(yīng)釜(上海貝倫儀器設(shè)備有限公司);DHG-9033A型電熱恒溫鼓風(fēng)干燥箱(沙鷹科學(xué)儀器有限公司);MD1444(8000-14000)型透析袋,上海源葉FD-IA-50型冷凍干燥機(jī)(北京博醫(yī)康實(shí)驗(yàn)儀器);T6新世紀(jì)型紫外可見分光光度計(jì)(北京普析通用儀器有限公司);Nicolet is50型傅立葉變換紅外光譜儀(賽默飛世爾科技有限公司);1658001型電泳儀設(shè)備(美國Bio-Rad公司);CHIRASCAN型圓二色光譜儀(英國Applied Photophysics);XRD(Spectris公司)。
1.2 方法
1.2.1人發(fā)角蛋白提取
按實(shí)驗(yàn)室已有方法提取人發(fā)角蛋白。稱取一定量人發(fā),剪至0.5 cm~1 cm,按1︰2︰2(w/v/v)的比例向燒杯中依次加入4% OP-10和5% NaOH,室溫下攪拌2 h。用去離子水沖洗6次,置于60℃烘箱過夜干燥。稱取2 g已預(yù)處理的人發(fā),置于200 mL水熱合成反應(yīng)釜內(nèi)膽內(nèi),加入8 mL 23%氨水,將反應(yīng)釜放入烘箱,185℃反應(yīng)30 min。加入50 mL去離子水,室溫下攪拌1.5 h,四號(hào)篩網(wǎng)過濾,收集濾液。濾液調(diào)pH至7.4,靜置后在4 ℃下12000 rpm離心20 min,合并上清液。上清液調(diào)pH至4.0~4.2,在4℃下6000 rpm離心20 min,留沉淀。角蛋白沉淀分散于1 mol·L-1碳酸氫鈉中,放入透析袋(截留分子量3500 Da)中透析,透析外液為去離子水,每12 h更換一次。
取不同透析時(shí)的角蛋白,冷凍干燥,得淺黃色粉末。
1.2.2聚丙烯酰胺凝膠電泳(SDS-PAGE)
參照Kadathur等[9]方法并稍作修改。配置12%的分離膠:1.6 mL去離子水、2 mL 30%丙烯酰胺溶液、1.3 mL 1.5 mol·L-1Tris(pH8.8)、0.05 mL 10% SDS溶液、0.05 mL 10%過硫酸銨和0.002 mL四甲基乙二胺。5%濃縮膠:2.1 mL去離子水、0.5 mL 30%丙烯酰胺混合溶液、0.38 mL 1.0 mol·L-1Tris(pH6.8)、0.03 mL 10% SDS溶液、0.03 mL 10%過硫酸銨和0.003 mL四甲基乙二胺。取20 μL角蛋白樣品(15 mg·mL-1)加入5×上樣緩沖液,煮沸10 min。取煮沸冷卻后的樣品10 μL和Marker 5 μL,上樣。80 V電壓下進(jìn)行電泳,待Marker條帶出現(xiàn)后,增大電壓至110 V,直至電泳分離結(jié)束。考馬斯亮藍(lán)R250染色,乙醇-醋酸脫色至無背景色。
1.2.3傅里葉紅外光譜(FT-IR)
將溴化鉀在紅外燈下于研缽中研磨成均勻粉末,再將角蛋白樣品與溴化鉀以1∶100的比例混合均勻后,研磨成粉。壓片機(jī)20 MPa壓制1 min~2 min成透明薄片,用FT-IR在400 cm-1~4000 cm-1掃描。
1.2.4接觸角
將角蛋白樣品壓片,置于接觸角測(cè)試儀平臺(tái)的玻璃片上,緩慢轉(zhuǎn)動(dòng)旋鈕,將水滴滴在在樣品上,記錄結(jié)果。
1.2.5二硫鍵含量的測(cè)定
參照Zhang等[10]方法。將角蛋白樣品各稱量10 mg。游離巰基含量測(cè)定:角蛋白樣品加入0.8 mL緩沖液A,室溫下震蕩2 h,加入0.2 mL緩沖液B,繼續(xù)震蕩1 h,在4 ℃下10000 rpm離心10 min。上清液稀釋至合適濃度,測(cè)量在412 nm的吸光度。空白參比為0.8 mL緩沖液A+0.2 mL緩沖液B。總巰基含量測(cè)定:參照游離巰基的測(cè)定方法。用緩沖液C和D替代緩沖液A和B。上述反應(yīng)在避光條件下進(jìn)行。計(jì)算時(shí),摩爾消光系數(shù)取13600 M-1·cm-1,游離巰基或總巰基含量計(jì)算公式為:

式中:A412為412 nm下角蛋白的吸光度值。二硫鍵含量計(jì)算公式為:
二硫鍵含量 =(總巰基含量-游離巰基含量)/2
1.2.6圓二色譜(CD)
參照Kadathur等[11]方法。用圓二色分光光度計(jì)對(duì)濃度為0.5 mg·mL-1的角蛋白樣品進(jìn)行圓二色性分析。25℃,以1 nm的光譜分辨率收集數(shù)據(jù),平均時(shí)間為1 s,掃描速率為100 nm·min-1,掃描范圍190 nm~250 nm,每個(gè)樣品重復(fù)測(cè)量三次。使用BeStSel服務(wù)器(http://bestsel.elte.hu)計(jì)算二級(jí)結(jié)構(gòu)。
1.2.7粉末X射線衍射(XRD)
取適量角蛋白樣品,研磨成細(xì)粉,過200目篩,進(jìn)行粉末X射線衍射測(cè)試。
1.3 數(shù)據(jù)處理
使用GraphPad軟件(Insight Science公司)進(jìn)行統(tǒng)計(jì)分析。顯著性水平設(shè)為α=0.05;用最小顯著性差異法檢驗(yàn)數(shù)據(jù)間的差異顯著性,用<0.05表示。
2 結(jié)果與討論
2.1 提取的人發(fā)角蛋白表征
2.1.1聚丙烯酰胺凝膠電泳(SDS-PAGE)
取透析了36 h所得的人發(fā)角蛋白,按照2.2.2方法,用SDS-PAGE測(cè)定其質(zhì)分子量,見圖1。
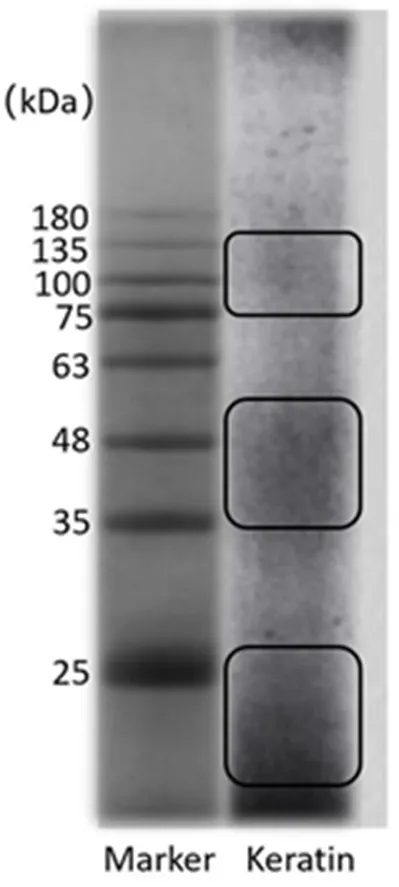
圖1 角蛋白的聚丙烯酰胺凝膠電泳圖
從圖1可以看出,提取所得角蛋白分布在25 kDa以下、35 kDa~56 kDa、75 kDa~135 kDa,分子量分布廣泛,與文獻(xiàn)基本一致[11-13]。
2.1.2傅里葉紅外光譜(FT-IR)
取透析了36 h所得的人發(fā)角蛋白,進(jìn)行FT-IR分析,并和人發(fā)纖維相比較,見圖2。

a.人發(fā)纖維;b.角蛋白
從圖2可以看出,天然人發(fā)纖維與提取的人發(fā)角蛋白在3200 cm-1~3400 cm-1均具有肽鍵酰胺A帶,1638 cm-1~1657 cm-1的酰胺I帶,1534 cm-1~1545 cm-1的酰胺II帶,1240 cm-1~1245 cm-1的酰胺III帶,說明兩者均具有典型的蛋白質(zhì)結(jié)構(gòu)[14,15],提取所得角蛋白與天然人發(fā)纖維分子結(jié)構(gòu)相同。
綜上可見,本文采用高溫水氨解自制的人發(fā)角蛋白,分子量分布為25 kDa以下、35 kDa~56 kDa、75 kDa~135 kDa,具有典型的角蛋白結(jié)構(gòu)。
2.2 透析時(shí)間對(duì)角蛋白的影響
角蛋白結(jié)構(gòu)是影響角蛋白應(yīng)用的重要因素[16]。因此,采用提取所得角蛋白,透析不同時(shí)間,研究對(duì)接觸角、二硫鍵和分子二級(jí)結(jié)構(gòu)的影響。
2.2.1接觸角
為了解角蛋白親水性的變化,采用接觸角測(cè)量來直觀定量。

圖3 角蛋白和人皮膚的接觸角圖
從圖3可以看出,透析時(shí)間的增加會(huì)使接觸角增大,角蛋白親水性降低。一般認(rèn)為,蛋白質(zhì)親水性的好壞主要取決于分子的表面結(jié)構(gòu),如果分子表面帶電氨基酸多,則親水性高,如果疏水殘基增多,則親水性變差。之所以會(huì)發(fā)生隨著透析時(shí)間增長(zhǎng),親水性變差,可能是因?yàn)殡S著透析時(shí)間增加,一些吸水性鹽被透析,或分子表面親水殘基內(nèi)卷。對(duì)水的親和能力極大地反映了角蛋白的應(yīng)用前景,正常皮膚的水接觸角為51.4°左右,過高或過低都不有利于其在生物修復(fù)材料中的應(yīng)用,36 h和72 h角蛋白的水接觸角都與正常皮膚的接觸角相差不大。
2.2.2二硫鍵
因角蛋白的提取過程往往涉及二硫鍵的斷裂,且二硫鍵在角蛋白結(jié)構(gòu)中起著重要作用,所以這里測(cè)量了二硫鍵含量變化。
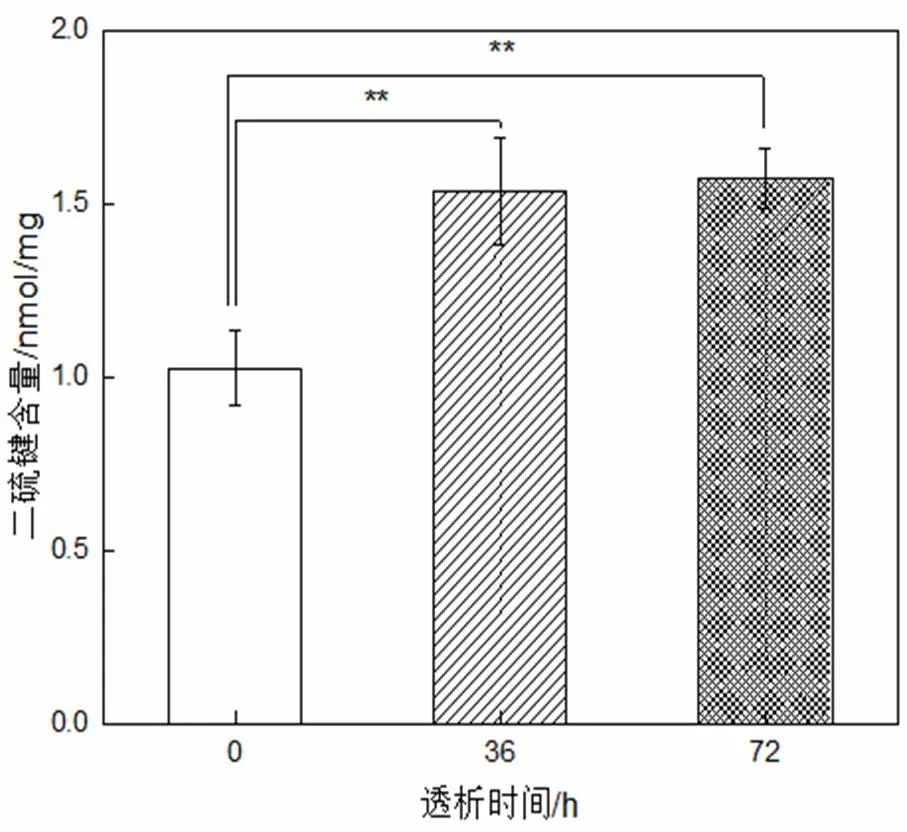
圖4 角蛋白的二硫鍵含量
從圖4可以看出,0 h~36 h,二硫鍵含量顯著增高;36 h~72 h,二硫鍵含量基本不變。這可能是因?yàn)榻堑鞍滋崛∩婕岸蜴I的斷裂,初始時(shí)未含二硫鍵的小分子量角蛋白較多,被透析至透析外液,36 h后幾乎透析干凈,故36 h~72 h二硫鍵含量基本不變。這也提示,高溫水氨解提取的人發(fā)角蛋白,透析提純可取36 h即可,而不用透析純化過長(zhǎng)時(shí)間[17-19]。
2.2.3二級(jí)結(jié)構(gòu)的影響
為了進(jìn)一步了解透析時(shí)間對(duì)角蛋白結(jié)構(gòu)的影響,用圓二色光譜(CD)對(duì)不同透析時(shí)間的角蛋白的二級(jí)結(jié)構(gòu)進(jìn)行了分析[21],見圖5。

圖5 不同透析時(shí)間角蛋白的二級(jí)結(jié)構(gòu)
蛋白質(zhì)空間結(jié)構(gòu)預(yù)測(cè)的第一步通常為預(yù)測(cè)二級(jí)結(jié)構(gòu),即判斷每一個(gè)氨基酸殘基是否處于α-螺旋、β-折疊和或其它狀態(tài)。每一段相鄰的氨基酸殘基具有形成一定二級(jí)結(jié)構(gòu)的傾向。從圖5(A)可以看出,不同角蛋白樣品均在200 nm~210 nm有一個(gè)負(fù)峰,可能是α-螺旋與無規(guī)則卷曲的復(fù)合峰;在222 nm有一個(gè)負(fù)峰,屬于α-螺旋。使用BeStSel服務(wù)器(http://bestsel.elte.hu)得到圖5(B),可以看出,在0 h時(shí),α-螺旋、β-折疊、無規(guī)則卷曲各占17.5%、38.6%、43.9%;在36 h時(shí),α-螺旋、β-折疊、無規(guī)則卷曲各占31.2%、37.6%、31.2%;在72 h時(shí),α-螺旋、β-折疊、無規(guī)則卷曲各占20.5%、12.0%、67.5%。
0~36 h透析過程中,α-螺旋含量增加,無規(guī)則卷曲減少,β-折疊含量幾乎未變,這可能是因?yàn)榍捌谕肝鲞^程中,無規(guī)則卷曲程度高的低分子量角蛋白擴(kuò)散至透析外液,使無規(guī)則卷曲減少,且α-螺旋結(jié)構(gòu)可能比β-折疊更穩(wěn)定,故α-螺旋含量顯著增高。
36 h~72 h透析過程中,無規(guī)則卷曲含量大幅度增加,可能說明在透析液中,無規(guī)則卷曲穩(wěn)定性最大,由于α-螺旋相比β-折疊又更穩(wěn)定,故在72 h后,無規(guī)則卷曲占比最大,α-螺旋次之,β-折疊最少。角蛋白的α-螺旋和β-折疊在長(zhǎng)時(shí)間透析中有向無規(guī)則卷曲靠攏的趨勢(shì)。結(jié)合圖3說明,無規(guī)則卷曲可能主要通過內(nèi)部的親水性基團(tuán)相互作用,從而使角蛋白表面疏水性增大,也變得更加穩(wěn)定。
2.2.4晶型的影響
圖6為角蛋白透析0 h、36 h、72 h和人發(fā)纖維的XRD圖譜。
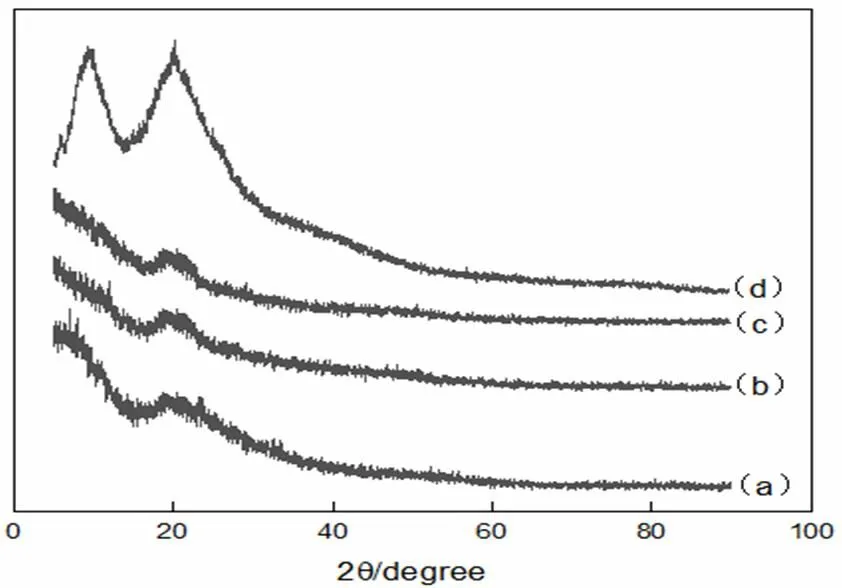
透析時(shí)間:a-0 h,b-36 h,c-72 h,d-人發(fā)纖維
在角蛋白的XRD圖譜中,9°和20°左右會(huì)存在兩個(gè)特征峰,9°的峰歸屬于α-螺旋和β-折疊,20°左右的峰則歸屬于β-折疊[20]。角蛋白提取物在9°和20°左右對(duì)應(yīng)的峰的尖銳程度都遠(yuǎn)低于人發(fā)纖維,表明晶體化程度低。從透析0 h,36 h和72 h的XRD圖可以看出,0 h和36 h的略有差別,主要表現(xiàn)在9°的峰形上,這可能是由于透析前的部分雜質(zhì)所引起;而36 h和72 h的XRD圖幾乎完全一致,結(jié)合圖5說明,α-螺旋或β-折疊向無規(guī)則卷曲變化過程中,α-螺旋和β-折疊晶區(qū)仍然會(huì)保留。
3 結(jié)論
本文提取人發(fā)角蛋白后,分別透析純化0 h、36 h、72 h。結(jié)果表明,隨著透析時(shí)間增加,角蛋白疏水性和二硫鍵均增加,透析至36 h后基本不變;隨著透析的進(jìn)行,α-螺旋和β-折疊,尤其是β-折疊易向無規(guī)則卷曲轉(zhuǎn)變;XRD結(jié)果顯示,這一轉(zhuǎn)變,對(duì)α-螺旋和β-折疊微晶區(qū)基本沒有影響。了解和掌握角蛋白二級(jí)結(jié)構(gòu)的變化情況,對(duì)角蛋白的生物應(yīng)用具有一定的指導(dǎo)意義。
[1] 袁小晶,尹海夢(mèng),樊曉瑋,等. 角蛋白/海藻酸鈉/聚丙烯酰胺水凝膠皮膚敷料的制備及創(chuàng)口修復(fù)研究[J]. 中國生物工程雜志,2021,41(8): 17-24.
[2] 王華杰,孫元元,盧亞楠,等. 肝素/人發(fā)角蛋白層層自組裝薄膜的細(xì)胞相容性及抗凝血性研究[C]. 河南省化學(xué)會(huì)2010年學(xué)術(shù)年會(huì)論文摘要集,2010.
[3] 竇潔. 基于一氧化氮釋放的角蛋白組織工程血管支架的制備研究[D]. 南京: 南京師范大學(xué),2021.
[4] Soodeh S, Jhamak N, Azadeh G, et al. Carboxymethyl cellulose-human hair keratin hydrogel with controlled clindamycin release as antibacterial wound dressing[J]. International Journal of Biological Macromolecules, 2020, 147: 1239-1247.
[5] Fereshteh A, Mohammad T Y, Abdolhadi F, et al. Keratin nanoparticles obtained from human hair for removal of crystal violet from aqueous solution: optimized by Taguchi method[J]. International Journal of Biological Macromolecules, 2020, 143: 492-500.
[6] Chen Y, Wang Y X. Keratin and its extraction[J]. Material Review, 2002, 16(12): 65-67.
[7] Azmi N A, Idris A, Yusof N S M. Ultrasonic technology for value added products from feather keratin[J]. Ultrasonics Sonochemistry, 2018, 47: 99-107.
[8] Cassoni A C, Freixo R, Pintado A I E, et al. Novel eco-fiendly method to extract keratin from hair[J]. ACS Sustainable Chemistry and Engineering, 2018, 6(9): 12268-12274.
[9] Kadathur R R, Ramar T, Balaraman M. Comparative analysis of the chemical treatments used in keratin extraction from red sheep's hair and the cell viability evaluations of this keratin for tissue engineering applications[J]. Process Biochemistry, 2020, 90: 223-232.
[10] Zhang Z, Nie Y, Zhang Q, et al. Quantitative change in disulfide bonds and microstructure variation of regenerated wool keratin from various ionic liquids[J]. ACS Sustainable Chemistry and Engineering, 2017, 5(3): 2614-2622.
[11] Jiang X U, Ping Z, Lin Z, et al. Study of keratin extraction from human hair[J]. Wool Textile Journal, 2015, 43(5): 38-42.
[12] Idris A, Vijayaraghavan R, Rana U A, et al. Dissolution of feather keratin in ionic liquids[J]. Green chemistry, 2013, 15(2): 525-534.
[13] Xu H, Yang Y. Controlled de-cross-linking and disentanglement of feather keratin for fiber preparation via a novel process[J]. Acs Sustainable Chemistry and Engineering, 2015, 2(6): 1404-1410.
[14] Aluigi A, Tonetti C, Rombaldoni F, et al. Keratins extracted from Merino wool and Brown Alpaca fibres as potential fillers for PLLA-based biocomposites[J]. Journal of Materials Science, 2014, 49(18): 6257-6269.
[15] Wang Y X, Cao X J. Extracting keratin from chicken feathers by using a hydrophobic ionic liquid[J]. Process Biochemistry, 2012, 47(5): 896-899.
[16] 錢迅南. 絲素蛋白/角蛋白復(fù)合材料的制備與性能研究[D]. 杭州: 浙江理工大學(xué),2020.
[17] 陳曉良. 人發(fā)角蛋白水凝膠對(duì)大鼠皮膚放射性復(fù)合傷修復(fù)促進(jìn)作用的研究[D]. 重慶: 重慶醫(yī)科大學(xué),2020.
[18] 徐江濤,朱平,張林,等. 人發(fā)角蛋白提取工藝研究[J].毛紡科技,2015,43(5): 38-42.
[19] Kelly S M, Jess T J, Price N C. How to study proteins by circular dichroism[J]. Biochimica et BiophysicaActa (BBA)-Proteins and Proteomics, 2005, 1751(2): 119-139.
[20] Huang T, Rui Y, Lin Z, et al. Programing performance of wool keratin and silk fibroin composite materials by mesoscopic molecular network reconstruction[J]. Advanced Functional Materials, 2016, 26(48): 9032-9043.
Study on Dialysis and Purification of Human Hair Keratin
In order to understand the changes of the secondary structure of human hair keratin during dialysis and purification, the human hair keratin extracted by high temperature ammonolysis was analyzed by contact angle analyzer, circular dichroism (CD) and X-ray diffraction (XRD). The results showed that during 0 h -36 h of dialysis, the hydrophilicity of keratin decreased, and the content of disulfide bond increased. The α-helix content increased significantly, and the proportion of random curl content decreased significantly. During 36 h -72 h of dialysis, the hydrophilicity still decreased, the disulfide bond content remained basically unchanged, and the proportion of irregular curl increased significantly. The dialysis process had little effect on α-helix and β-folded crystal region.
human hair keratin; dialysis; purification; time; structure
Q518.1
A
1008-1151(2022)08-0043-04
2022-06-08
郭克兵(1996-),女,河北邢臺(tái)人,重慶大學(xué)化學(xué)化工學(xué)院學(xué)生,研究方向?yàn)榻堑鞍滋崛C(jī)理。
季金茍(1962-),男,江西南昌人,重慶大學(xué)化學(xué)化工學(xué)院教授,研究方向?yàn)樯锊牧稀?/p>
猜你喜歡
小獼猴智力畫刊(2023年4期)2023-04-23 08:49:58
哲學(xué)評(píng)論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
中學(xué)生數(shù)理化·高一版(2018年1期)2018-02-10 05:20:03
影視與戲劇評(píng)論(2016年0期)2016-11-23 05:26:01
七彩語文·寫字與書法(2016年7期)2016-07-28 21:40:22
七彩語文·寫字與書法(2016年6期)2016-07-15 19:36:34
人間(2015年21期)2015-03-11 15:23:21
現(xiàn)代企業(yè)(2015年9期)2015-02-28 18:56:50
