樹枝狀介孔二氧化硅納米球負載氧化鈷催化劑催化氧化甲苯
2022-09-13 12:53:54趙秋娜羅明生劉清龍
石油學報(石油加工) 2022年5期
趙秋娜, 羅明生,3, 劉清龍, 楊 智
(1.北京石油化工學院 新材料與化工學院,北京 102617;2.北京工業大學 環境與生命學部,北京 100124;3.北京石油化工學院 燃料清潔化及高效催化減排技術北京市重點實驗室,北京 102617)
大多數揮發性有機化合物(VOCs)對環境和人類健康有害[1-2]。控制VOCs排放的技術眾多,如吸附、吸收、催化燃燒、光催化氧化和催化氧化等[3-5]。催化氧化方法由于其高效率、低成本且無二次污染[6-7],被認為是減少VOCs排放最有應用前景的途徑之一。VOCs催化氧化的核心問題是高效催化劑的開發,其中單一和復合過渡金屬氧化物[7-9]、負載型貴金屬或過渡金屬氧化物[10-12]等催化劑被廣泛用于VOCs催化氧化應用與研究。
過渡金屬氧化物(如Co3O4[13-14]、MnO2[2]、LaFeO3[15]等)含有晶格缺陷(氧空位)和多種氧化態金屬離子,氧化過程中形成的氧化-還原循環有利于甲苯的吸附和活化,在VOCs去除中表現出良好的催化活性,一直是研究的熱點。例如具有獨特雜化結構的α-MnO2/Mn3O4[5]因豐富的活性界面表現出優異的低溫催化性能,可與部分貴金屬基催化劑相媲美[16-17]。α-MnO2/Mn3O4對甲苯的轉化率為90%時,α-MnO2和Mn3O4分別僅約為70.6%和7.6%,并且在單位時間內可生成更多的超氧自由基,其甲苯轉化率在30 h內可以保持在95%以上。過渡金屬復合氧化物間相互作用在VOCs消除中發揮重要作用。而鈷氧化物具有多種氧化態和晶格氧缺陷,與氧結合速率較高,廣泛應用于甲苯催化氧化反應中,表現出良好的催化性能。Bai等[18]通過水熱或微乳化途徑制備了一系列Co3O4催化劑,以十六烷基三甲基溴化銨(CTAB)為表面活性劑所制得的Co3O4催化劑具有更低的表面Co3+/Co2+摩爾比、更高的表面吸附氧濃度和更好的低溫還原性,表現出良好的甲苯氧化催化性能。而將氧化鈷做成多孔材料可以提高其傳質效果和活性位的接觸性,進而改善催化劑的催化性能。例如使用SBA-15及KIT-6介孔氧化硅作為硬模板,通過納米澆鑄的方法把氧化鈷做成有序介孔材料,可以提高體相氧化鈷的比表面積、Co2+含量和表面活性氧物種含量,減少擴散阻力,從而提高催化劑的催化性能[19]。Fang等[20]基于ZIFs材料作為模板成功地制備了核-殼結構的CeO2@Co3O4催化劑,該催化劑表面呈分層皺紋形態,這種獨特的結構,尤其是核與殼之間的界面使CeO2@Co3O4催化劑具有優異的甲苯氧化催化性能,且XPS和H2-TPR結果表明,雙金屬催化劑中鈷和鈰的氧化物之間存在協同作用,提高了催化劑的還原性和活性。
負載型催化劑是一類重要的催化劑體系,作為催化劑的一個重要組成部分,載體可增加催化劑的比表面積,提高活性金屬的分散度,改善催化劑的反應性能。例如常見的不可還原的氧化物(Al2O3、SiO2、MgO、ZnO和La2O3)和可還原的氧化物(Co3O4、CeO2、TiO2和Fe2O3)載體[21],載體的差異會影響負載型鈷基催化劑的性能。Luo等[22]制備的負載型CoOx催化劑中,與SBA-15、γ-Al2O3和AC載體相比,石墨化的氮化碳(g-C3N4)載體富含N和電子,具有更佳的催化性能。具有高表面積、規則介孔結構和高穩定性的各種介孔材料有利于活性組分的分散和反應氣體的擴散。例如在消除甲苯反應中,與體相Co3O4樣品相比,三維有序大孔結構的Co3O4/Eu0.6Sr0.4FeO3(Co3O4/ESFO)樣品表現出更好的催化活性[23],甲苯轉化率為90%時的溫度(T90)達到279 ℃,這與其較高的氧物種濃度、更好的低溫還原性以及Co3O4與3DOM-ESFO間相互作用密切相關。Szegedi等[24]制備了Co/MCM-41和Co/SBA-15催化劑,研究其在甲苯氧化中的催化性能,發現浸漬溶液的pH值影響了介孔載體的表面電荷和氧化鈷與載體間的相互作用,形成不同的鈷氧化物種(Co3O4、Co2+、Co-硅酸鹽類物質),其中形成的Co3O4物種易還原且高度分散,具有最高的催化活性。最近,文獻[25-27]報道了一種新型樹枝狀介孔二氧化硅納米球(DMSNs)材料,該材料具有獨特的中心徑向孔結構,有望成為催化劑的新型載體。且具有較大的孔道入口和均勻的單分散納米顆粒結構,與常見的HMS、MCM-41和SBA-15等介孔二氧化硅材料相比具有獨特優勢。該載體不易被堵塞,并且具有良好的活性位點可及性和反應物的快速擴散性。此外,常見的介孔氧化硅材料的合成過程周期較長,通常需要幾天的時間,而DMSNs材料制備周期較短,可在數小時內完成合成,可以通過摻雜活性異質元素對該納米材料進行功能化和修飾改性。
筆者以樹枝狀介孔二氧化硅納米球(DMSNs)材料為載體,通過等體積浸漬法制備了一系列不同氧化鈷負載量的DMSNs催化劑(Co/DMSNs),利用多種表征手段對該催化劑的理化結構、氧化還原性、表面元素組成、催化劑活性氧物種等特性進行了系統分析,并考察了該催化劑催化氧化甲苯的性能及催化劑的穩定性。
1 實驗部分
1.1 原料和試劑
三乙醇胺、CTAB、水楊酸鈉、原硅酸四乙酯、六水合氯化鈷,均為分析純,北京伊諾凱科技有限公司產品。
1.2 催化劑制備
1.2.1 DMSNs介孔納米球載體的制備
使用陽離子型表面活性劑CTAB和水楊酸鈉作為結構導向劑,原硅酸四乙酯作為硅源,三乙醇胺作為合成過程的催化劑,通過水熱合成法,合成了樹枝狀介孔二氧化硅納米球(DMSNs)載體[26]。合成過程如下:首先稱取0.41 g三乙醇胺放入燒杯中,向其中加入150 mL蒸餾水,將燒杯放入80 ℃水浴中,恒溫攪拌0.5 h。再向燒杯中加入2.28 g十六烷基三甲基溴化銨及1.01 g水楊酸鈉,繼續恒溫攪拌1 h。向燒杯緩慢加入22.43 g原硅酸四乙酯,繼續恒溫攪拌2 h。將燒杯中得到的混合液倒入晶化釜并置于80 ℃烘箱中靜置4 h,隨后自然冷卻至室溫,對沉淀物進行抽濾并洗滌數次后,再置于80 ℃烘箱中干燥12 h。最后放入馬弗爐中進行焙燒,從室溫經過5 h勻速(升溫速率為1.75 ℃/min)升至550 ℃,在550 ℃下保持6 h得到固體粉末,記為DMSNs。
1.2.2 等體積浸漬法合成Co/DMSNs催化劑
將一定量的六水合氯化鈷(CoCl2·6H2O)溶解到去離子水中得到浸漬溶液。將浸漬溶液逐滴加入到干燥的DMSNs載體中,邊滴加邊用玻璃棒攪拌。將攪拌均勻的浸漬樣品進行超聲震蕩處理0.5 h,隨后在60 ℃下干燥12 h,最后放入馬弗爐中進行焙燒,在400 ℃下保持6 h得到Co/DMSNs催化劑。通過改變氯化鈷的添加量,分別獲得鈷負載量(質量分數,下同)為0.2%、0.5%、1.7%、3%、5%、10%、20%和30%的催化劑,分別命名為0.2%Co/DMSNs、0.5%Co/DMSNs、1.7%Co/DMSNs、3%Co/DMSNs、5%Co/DMSNs、10%Co/DMSNs、20%Co/DMSNs、30%Co/DMSNs。
1.3 催化劑表征
在日本島津XRD-7000型X射線衍射儀(XRD)上測定樣品物相結構,CuKα射線(λ=0.15418 nm),操作電壓為40 kV,電流為30 mA,掃描范圍為10°~90°,掃描速率為4°/min。
采用美國QUANTE400F型掃描電子顯微鏡(SEM)和JEM-2100型透射電子顯微鏡(TEM)進行樣品的形貌觀察。
使用日本BELSORP-max型物理吸/脫附儀測定催化劑的孔結構,將樣品在300 ℃的條件下真空干燥4 h后,在液氮中冷卻至-196 ℃,進行低溫N2吸附-脫附實驗,比表面采用Brunauer-Emmet-Teller(BET)方法計算得出,孔結構根據BJH模型測得。
采用日本BELCAT-B型全自動程序升溫化學吸附儀測定催化劑氧化還原性(H2-TPR)。將約50 mg的催化劑置于石英反應管中,在高純Ar(30 mL/min)下于300 ℃預處理1 h,然后冷卻至50 ℃開始還原過程,在10%(體積分數)H2/Ar氣氛下以10 ℃/min升溫到900 ℃,用TCD檢測器記錄氫氣消耗量。
在與H2-TPR相同的裝置上進行氧氣程序升溫脫附(O2-TPD)。在進行O2-TPD實驗之前,將30 mg的樣品在30 mL/min N2氣流中及300 ℃下預處理1 h。樣品冷卻至室溫后,在40 mL/min的O2氣氛中吸附1 h,然后使用30 mL/min的He氣吹掃1 h,從而清除掉系統中殘留的O2。將樣品以10 ℃/min的升溫速率從室溫加熱到800 ℃,與此同時記錄O2解吸信號。
采用日本島津Kratos AXIS Ultra DLD X射線光電子能譜分析儀(XPS)測定樣品表面的元素組成和化學狀態。激發源為單色化AlKα射線(hυ=1486.6 eV),功率為45 W,工作電壓為15 kV,發射電流為3 mA。
用電感耦合等離子體光發射光譜法(ICP-OES)在Agilent 725儀器上測定Co負載量。
采用Renishaw inVia共聚焦拉曼光譜儀測定樣品的拉曼光譜,激發光波長為532 nm。
在JEOL JES-FA 100 EPR光譜儀上通過電子順磁共振探測樣品表面氧空位。光譜儀在X波段上使用標準TE011圓柱諧振器工作。可變溫度控制器ES-DVT4可在溫度(-150~150 ℃)下檢測EPR光譜。將由EPR光譜儀數據系統計算機控制溫度下的冷卻氣體(液氮)供入樣品區域,獲得所需溫度。
1.4 催化劑反應性能評價
在微型反應裝置上進行催化氧化甲苯的反應。將一定量的催化劑壓片后研磨篩分出40~60目的顆粒,置入石英反應管(直徑6 mm)中部,反應管底部放入石英棉用于承托催化劑。采用連續流進樣,50 μL/L的甲苯+20.0%(所占氣體的體積分數)O2+N2(平衡氣)在混合罐中混合后進入反應管,總體積流量為50 mL/min。反應前,將混合氣連續通入空管,獲得穩定初始濃度。然后將0.2 g催化劑置于具有石英棉的反應管中,在給定溫度下待穩定0.5 h后進行尾氣分析。反應尾氣采用天美GC7900氣相色譜儀進行分析,檢測條件為: TM-5毛細色譜柱(15 m×0.25 mm×0.25 μm),高純氬氣為載氣,FID檢測器。反應條件為:常壓,質量空速15000 mL/(g·h)。
以甲苯的轉化率(X,%)作為評價催化劑性能的指標,具體計算公式見式(1)。
X=(φin-φout)/φin×100%
(1)
式中:φin為進入反應器前甲苯的初始體積分數,μL/L;φout為反應尾氣中甲苯的體積分數,μL/L。
通過活性評價反應裝置考察了水蒸氣對催化劑性能的影響。控制水蒸氣的體積分數為1%,利用恒溫水浴鍋保持水蒸氣穩定在40 ℃,然后通過調節載氣(N2)的流速控制水蒸氣的濃度,流速的計算公式見式(2)。
pw/p0=φw×10-6×(FT/FN2)
(2)
式中:pw為水溶液在40 ℃時的飽和蒸汽壓,pw=7.381 kPa;p0為標準大氣壓,p0=101.325 kPa;φw為水蒸氣的體積分數,μL/L;FT為混合氣體的總體積流量,mL/min;FN2為攜帶水蒸氣的氮氣體積流量,mL/min。
2 結果與討論
2.1 催化劑表征結果分析
2.1.1 XRD表征
圖1為不同鈷負載量的Co/DMSNs催化劑的XRD譜圖。圖1中位于20°~30°范圍內的寬峰歸屬于無定形SiO2的特征衍射峰,可知合成的催化劑載體為無定型非晶體結構。在Co負載量較低(0.2%~0.5%)時,沒有出現明顯歸屬于鈷氧化物的特征峰,這說明鈷氧化物在DMSNs載體表面上的分散度相對較高,并沒有聚集形成比較大的體相顆粒,已超過XRD的檢測限;或DMSNs表面上的鈷氧化物可能是以無定型結構存在。隨著Co負載量的不斷提高,Co/DMSNs催化劑在2θ為19.0°、31.2°、36.8°、44.8°、59.3°和65.2°處出現明顯的特征峰,經與標準卡片對比,歸屬于Co3O4的特征衍射峰(PDF#42-1467)[22];對應的晶面分別為(111)、(220)、(311)、(400)、(511)、(440)晶面,表明催化劑中出現了Co3O4晶相物種。相應的Co3O4特征峰的強度隨著Co負載量的提高而增強,說明Co負載量的提高導致鈷氧化物不斷聚集,形成Co3O4晶相物種。此外,Co負載量高于10%的催化劑中出現了六水合氯化鈷的衍射峰,說明當氯化鈷負載量較高時,所合成的催化劑在焙燒過程不易完全被氧化成鈷氧化物。特別是以無定形SiO2為載體的催化劑中,六水合氯化鈷的衍射峰更為明顯,這可能是因為無定形SiO2載體不利于氯化鈷的分散,在焙燒過程中造成氯化鈷的殘留。
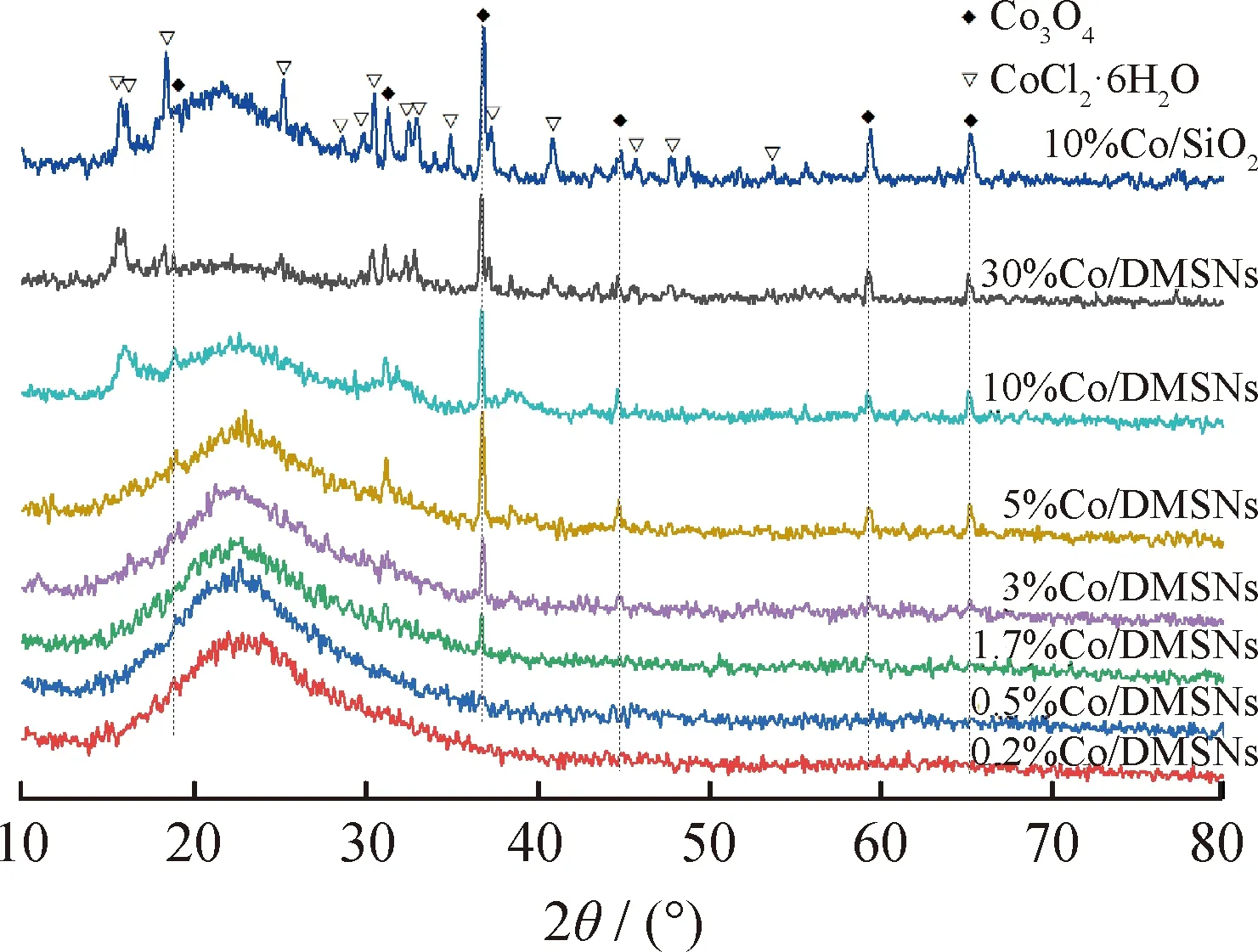
圖1 不同鈷負載量Co/DMSNs催化劑的XRD譜圖Fig.1 XRD patterns of Co/DMSNs catalysts with different cobalt loadings
2.1.2 SEM及TEM表征
通過TEM、HRTEM和SEM技術進一步表征了不同鈷負載量Co/DMSNs催化劑的微觀形貌及催化劑中鈷氧化物的分散性,結果如圖2(SEM照片)和圖3(TEM和HRTEM照片)所示。由圖2可以看到,所有Co/DMSNs催化劑呈現出樹枝狀納米球的結構形態,表面是褶皺的SiO2薄片,納米球直徑約為280 nm。圖3顯示該納米球內部是中心-徑向的孔結構,孔徑逐漸變寬,形成了約35 nm的徑向大孔開口。中心-徑向的介孔結構能夠有效地促進鈷氧物種的分散,進而抑制其在催化劑表面的聚集或者堵塞介孔孔道。隨著Co含量的增加,樣品保持了DMSNs載體的樹枝狀介孔納米球的形態結構,但Co負載量較高(30%,圖2(f))時,部分樹枝狀的褶皺表面形態受到一定影響,原來均勻的納米球從單分散的顆粒變為交聯的團簇。此外,當催化劑樣品中Co負載量超過5%后,從HRTEM照片(圖3(c)、(f)和(i))中可以觀察到少量氧化鈷顆粒的存在,粒徑為1~2 nm,表明氧化鈷在載體上分散度較高,沒有聚集形成較大的體相氧化鈷顆粒。

圖2 不同鈷負載量Co/DMSNs催化劑的SEM照片Fig.2 SEM images of Co/DMSNs catalysts with different cobalt loadings(a) 0.5%Co/DMSNs; (b) 1.7%Co/DMSNs; (c) 3%Co/DMSNs; (d) 5%Co/DMSNs; (e) 10%Co/DMSNs; (f) 30%Co/DMSNs

圖3 不同鈷負載量Co/DMSNs催化劑的TEM及HRTEM照片Fig.3 TEM and HRTEM images of Co/DMSNs catalysts with different cobalt loadings(a)—(c) 1.7%Co/DMSNs; (d)—(f) 5%Co/DMSNs; (g)—(i) 10%Co/DMSNs
2.1.3 N2物理吸附-脫附表征
圖4為Co/DMSNs催化劑樣品的N2吸附-脫附等溫線和孔徑分布。從圖4可以看出,所有樣品的吸附-脫附等溫曲線的變化趨勢基本相同,均為典型的Ⅳ型等溫線(IUPAC分類)且具有H3型滯后環特征,無明顯的飽和吸附平臺,說明合成的催化劑樣品均為典型的有序介孔材料[20],具有較好的孔結構。Co負載量對催化劑的孔徑分布影響較小,制備的催化劑仍均為介孔材料,孔徑主要分布在10~60 nm之間,在25 nm和35 nm左右出現2個明顯的分布峰。
表1為不同Co負載量的Co/DMSNs催化劑的N2物理吸附-脫附測試分析結果。由表1可知,隨著Co負載量的增加,Co/DMSNs催化劑的比表面積和總孔體積均呈現下降趨勢,而其平均孔徑變化不大。其中,當Co負載量為0.5%~10%時,催化劑孔體積下降不明顯,通過SEM結果可以看到高載量的樣品有些破損,出現一定的黏連和重構。當Co負載量增加至30%時,所制備的催化劑比表面積由426 m2/g降至165 m2/g,總孔體積從1.45 cm3/g降至0.71 cm3/g,比表面積和孔體積下降十分明顯。這說明隨著Co負載量的提高,催化劑載體表面上的褶皺或孔道逐漸被填充,引起催化劑比表面積和孔體積的下降,但是與介孔孔徑較小的介孔材料(例如MCM-41、SBA-15等)相比[24,28],其比表面積下降趨勢相對緩慢。此外,催化劑的平均孔徑仍保持在33~38 nm之間,與孔徑分布圖的結果一致。這表明Co的加入沒有顯著影響到催化劑的孔道特性,DMSNs載體特殊的中心放射狀的孔道結構更有利于活性組分的分散而不易堵塞孔道,能夠較好地保持載體原來的有序介孔結構。因此,Co/DMSNs催化劑較大的比表面積和孔徑,不僅為甲苯氧化反應提供較高的接觸面積和活性位點,也有利于反應物和產物在孔道內的擴散。

表1 不同鈷負載量Co/DMSNs催化劑的結構特性和實際金屬負載量Table 1 Structural characteristics and actual metal loading of Co/DMSNs catalysts with different cobalt loadings

圖4 不同鈷負載量Co/DMSNs催化劑的N2吸附-脫附等溫線和孔徑分布Fig.4 N2 adsorption-desorption isotherms and pore size distribution of Co/DMSNs catalysts with different cobalt loadings(a) N2 adsorption-desorption isotherms; (b) Pore size distribution
2.1.4 H2-TPR表征
用H2-TPR進一步表征了Co/DMSNs催化劑的氧化還原性能,結果如圖5所示。由圖5可見,不同Co負載量的Co/DMSNs催化劑的還原峰位置具有明顯的差異。當Co負載量低于10%時,在300~400 ℃之間存在2個還原峰,分別歸屬于Co3+→Co2+和Co2+→金屬Co的還原過程。在Co負載量較低(<3%)時,在500~800 ℃之間出現了還原峰,這表明催化劑中的鈷氧化物與載體之間相互作用較強,這可能是由于在焙燒過程中形成了較難還原的鈷硅物種[29]。當Co負載量增加(>3%)時,在460~480 ℃之間出現1個明顯的還原峰,這可能是高分散的納米小顆粒鈷氧物種的還原過程[30]。而當Co負載量提高到30%時,催化劑的還原峰發生疊加形成了1個包峰,這歸屬于體相Co3O4的還原峰。通過H2-TPR表征結果可知,在Co負載量較低(<1.7%)時,催化劑中CoOx物種的分散度相對較高,并且部分與氧化硅載體發生較強的相互作用。當Co負載量提高到1.7%以上時,還原曲線中出現了尖銳的還原峰,表明催化劑中出現了顆粒狀的鈷氧化物,這與XRD的表征結果一致。此外,Co2+的還原峰面積明顯高于Co3+的還原峰面積,表明催化劑中本身存在大量Co2+物種,相應的XPS表征結果顯示Co2+/Co3+摩爾比大于2,也證明催化劑中存在大量的Co2+物種。但是Co負載量進一步提高到30%時,過高的Co負載量不僅影響載體的形貌結構,而且逐漸堵塞了介孔孔道,形成了大量的體相氧化鈷物種,最終其還原過程與純體相氧化鈷基本相似[31]。
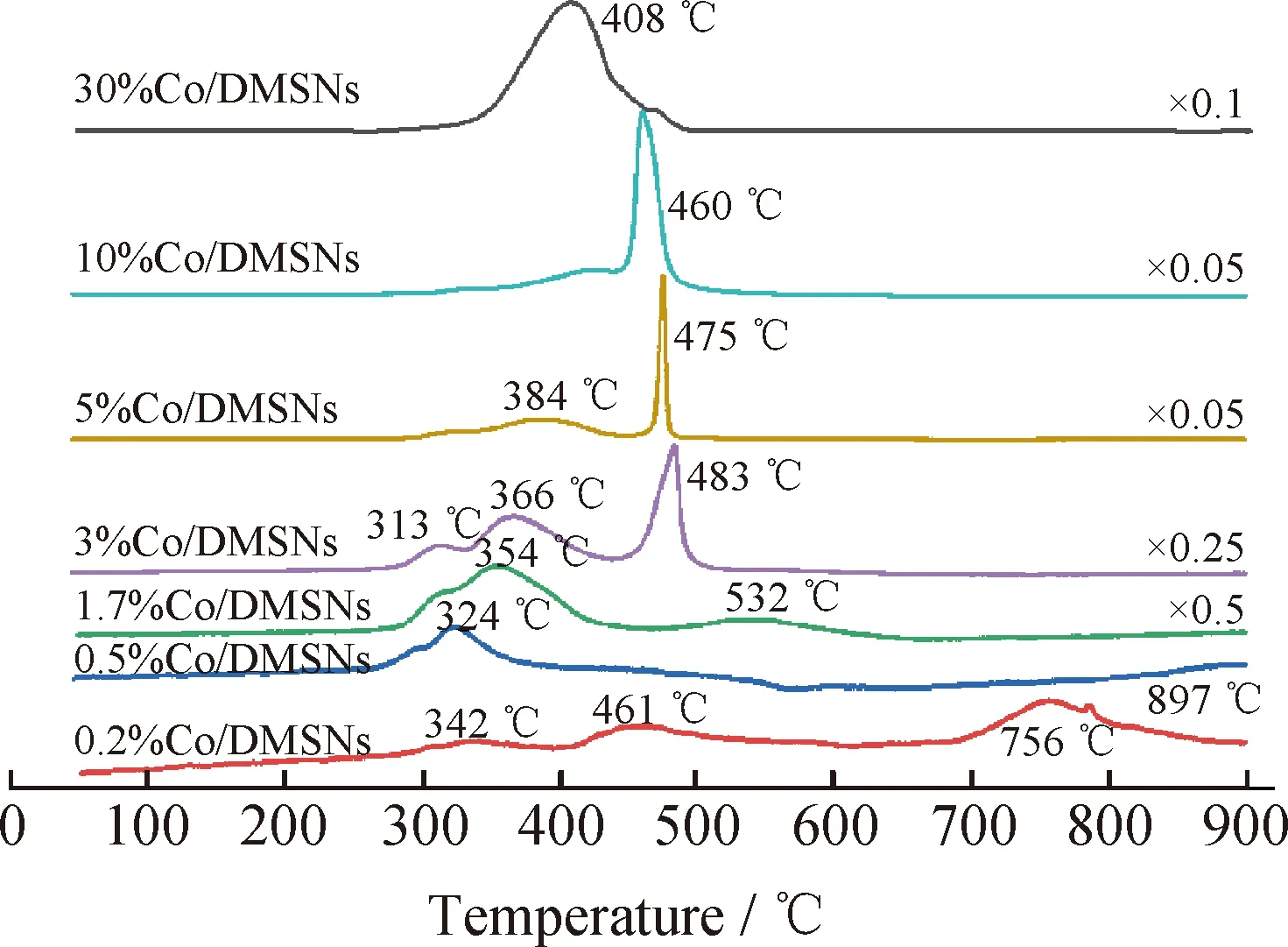
The intensity of some profiles is reduced for clarity(e.g. ×0.5 represents that the intensity in the figure is half of actual intensity).圖5 不同鈷負載量Co/DMSNs催化劑的H2-TPR曲線Fig.5 H2-TPR curves of Co/DMSNs catalysts with different cobalt loadings
2.1.5 O2-TPD表征
反應物在催化劑表面的吸附是發生多相催化反應的關鍵,氧氣分子的吸附性能是影響催化劑活性的重要因素,因此采用氧氣程序升溫脫附(O2-TPD)實驗研究了Co/DMSNs催化劑表面與吸附氧物種之間的相互作用。圖6為不同鈷負載量Co/DMSNs催化劑的O2-TPD圖譜。催化劑中通常存在4種不同類型的氧物種:吸附在氧空位上的分子氧物種(Ov)、吸附在表面的氧離子(Oads)、表面晶格氧(Olatt,s)和體相中晶格氧(Olatt,b)[32]。在程序升溫過程中,解吸能力由強到弱的順序依次遵循Ov、Oads、Olatt,s、Olatt,b[33]。如圖6所示,解吸峰大致分為70~120 ℃、120~450 ℃、450~700 ℃和>700 ℃ 4段溫度區間,分別對應于吸附在氧空位上的分子氧物種(Ov)、表面吸附氧離子(Oads)、表面的晶格氧和體相的晶格氧。隨著Co負載量的提高,10%Co/DMSNs催化劑在較低溫度下出現了較大的氧脫附峰,其峰面積明顯大于其他催化劑的低溫脫附峰面積,表明10%Co/DMSNs樣品具有更豐富的表面活性氧物種。
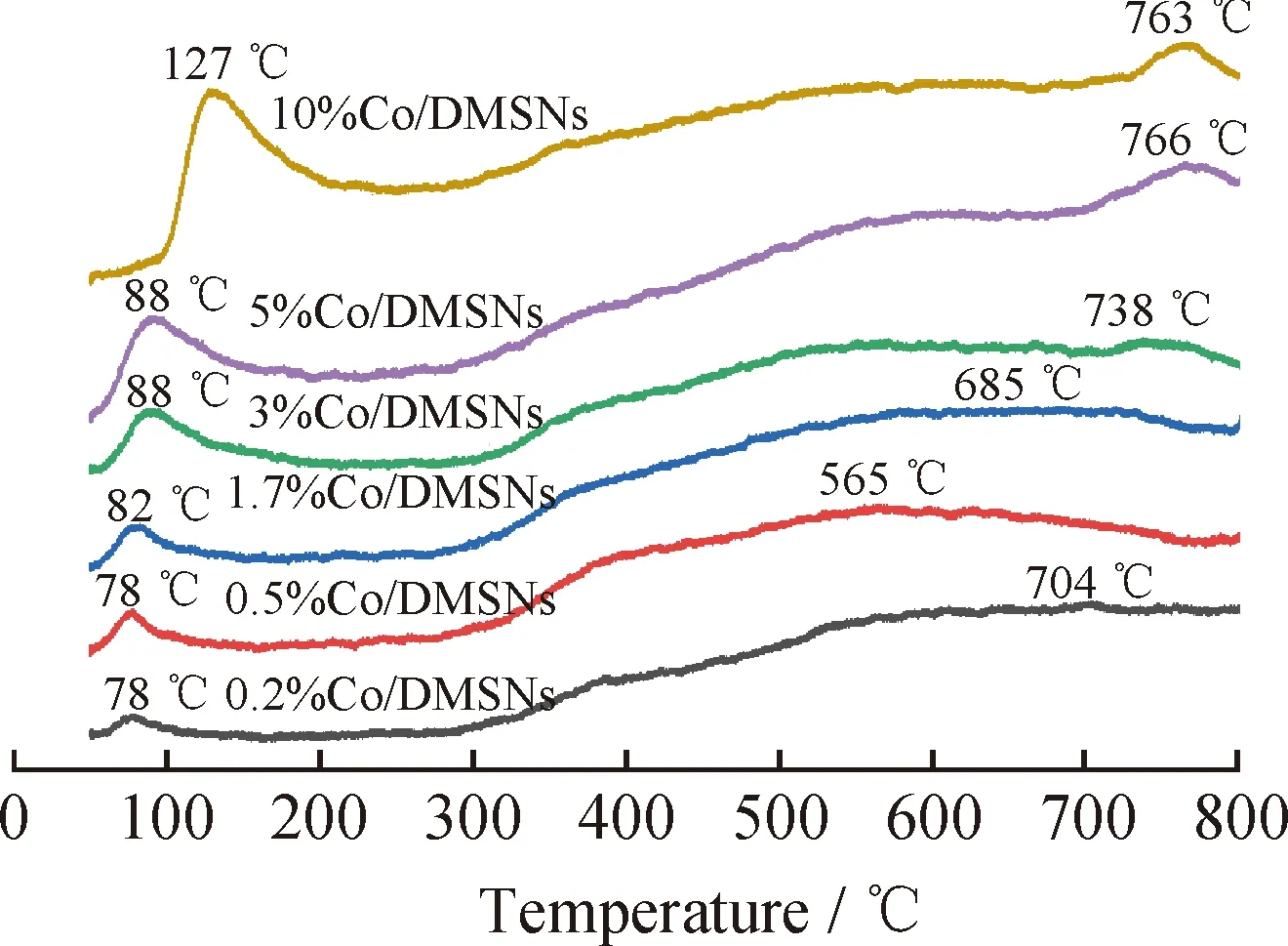
圖6 不同鈷負載量Co/DMSNs催化劑的O2-TPD圖譜Fig.6 O2-TPD spectra of Co/DMSNs catalysts with different cobalt loadings
2.1.6 XPS表征
催化劑表面Co的價態對其催化性能具有重要影響,因此利用XPS光譜對部分催化劑近表面的化學狀態進行了進一步表征。圖7為部分代表性Co/DMSNs催化劑的Co 2p光譜圖。Co 2p光譜圖由兩組峰組成,分別為Co 2p3/2和Co 2p1/2特征峰。由圖7可知,在Co 2p譜圖中,催化劑的Co 2p1/2的結合能在798 eV左右,而Co 2p3/2的結合能位于782 eV左右。將Co 2p3/2譜峰進行分峰處理,其中,結合能在779.7 eV左右的擬合峰歸屬于Co3+的特征峰,在781.7 eV左右的擬合峰可歸屬于Co2+的特征峰[34]。通過對二者的特征峰進行峰面積積分,可得到不同催化劑中Co2+/Co3+的摩爾比。結果發現,隨著Co負載量的提高,催化劑中Co2+/Co3+的摩爾比逐漸升高,10%Co/DMSNs催化劑較3%Co/DMSNs 和1.7%Co/DMSNs催化劑具有明顯更高的Co2+/Co3+摩爾比,可達到7左右。這表明10%Co/DMSNs催化劑中的Co主要是以Co2+的形式存在。此外,在786.3 eV左右存在衛星峰,表明鈷氧化物與載體間存在相互作用[35],特別是10%Co/DMSNs催化劑中CoOx物種與載體之間存在較強的相互作用,這對催化劑的穩定性具有重要影響。
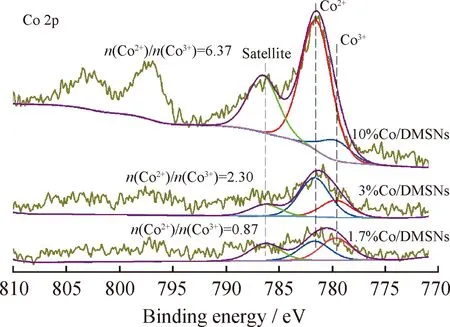
圖7 不同鈷負載量Co/DMSNs催化劑的Co 2p XPS譜圖Fig.7 Co 2p XPS spectra of Co/DMSNs catalysts with different cobalt loadings
2.1.7 Raman表征
拉曼光譜是研究缺陷的有力工具,它對材料的晶格畸變非常敏感。圖8為不同鈷負載量Co/DMSNs催化劑的拉曼譜圖。由圖8可知,各個不同Co負載量的催化劑均在195、481、520、619和690 cm-1處出現5個顯著的振動峰,分別歸屬于尖晶石Co3O4的四面體CoO4的F2g(1)、E2g、F2g(2)、F2g(3)對稱和八面體CoO6的A1g對稱[14,36]。當Co負載量超過10%時,歸屬于尖晶石Co3O4的拉曼特征峰明顯增長,這說明催化劑中形成大量的Co3O4物種。而10%Co/DMSNs催化劑雖然也出現一定量的Co3O4物種,但是拉曼特征峰相對較低,說明催化劑中的鈷氧物種主要還是以高分散或無定型的鈷氧物種形式存在。一般來講,紅移的產生表明催化劑有高度的缺陷結構,這將有利于將吸附的氧分子活化成活性氧。與10%Co/DMSNs催化劑相比,其他Co負載量相對低的催化劑的拉曼振動峰向高波數方向偏移,說明10%Co/DMSNs具有更多的結構缺陷。拉曼光譜結果表明,10%Co/DMSNs具有更多的結構缺陷構成的活化中心,氧分子可以吸附在這些缺陷位上形成活性氧物種,從而促進甲苯的催化氧化。
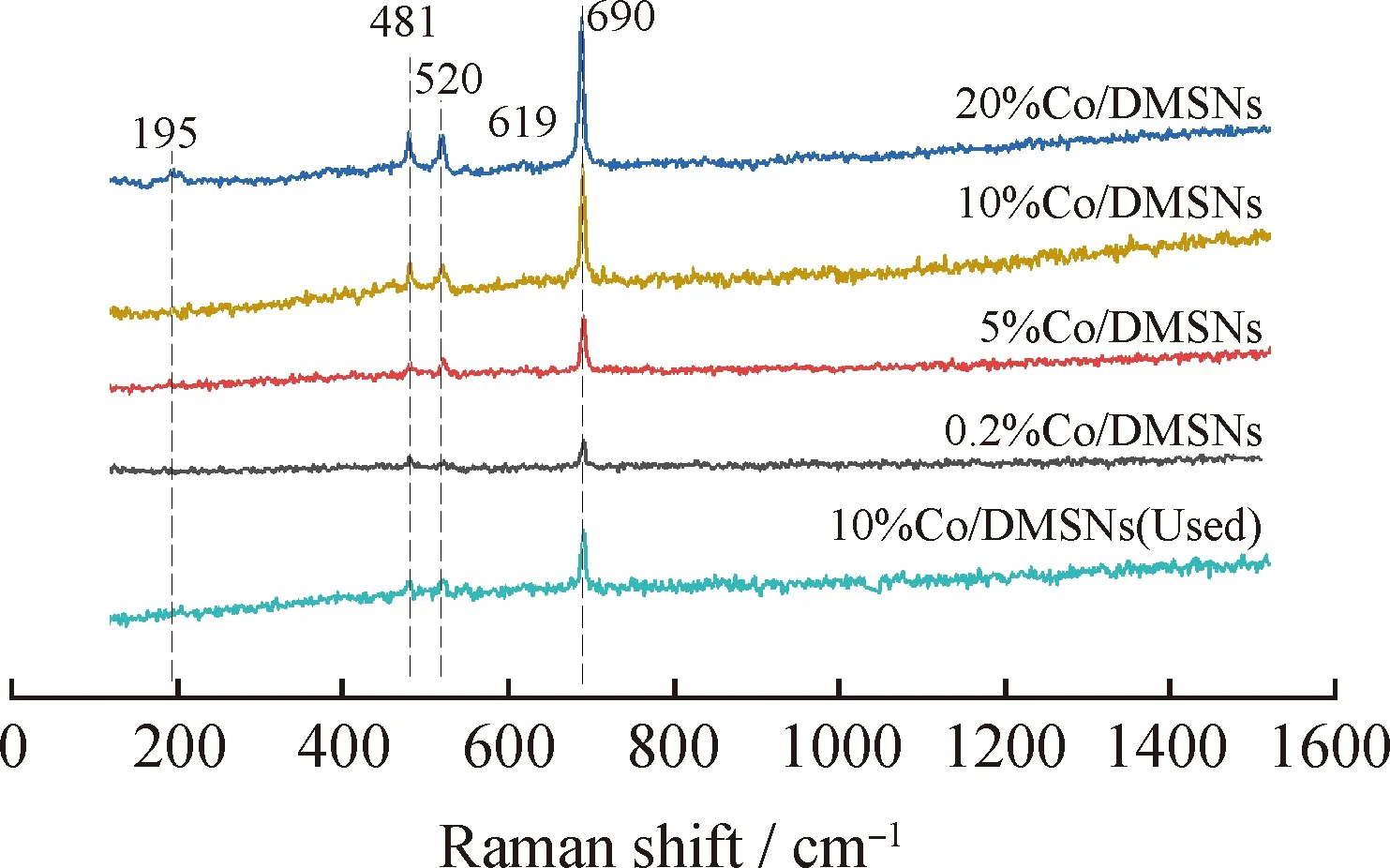
圖8 不同鈷負載量Co/DMSNs催化劑的拉曼譜圖Fig.8 Raman spectra of Co/DMSNs catalysts with different cobalt loadings
2.1.8 EPR表征
為了進一步證實催化劑表面氧空位的存在,對部分催化劑進行了電子順磁共振(EPR)的表征分析,結果如圖9所示。每個樣本都有一個對稱的EPR,歸因于納米材料氧空位處的未配對電子,信號強度可以反映氧空位的濃度[37]。從圖9可以看出,10%Co/DMSNs催化劑的峰值強度稍高于1.7%Co/DMSNs催化劑的峰強,表明10%Co/DMSNs催化劑內部晶格中存在更多的氧空位,這有利于提高其催化氧化性能。但整體而言,2個催化劑的EPR譜圖區別不大,說明氧空位數量不是影響該系列催化劑活性的關鍵因素。
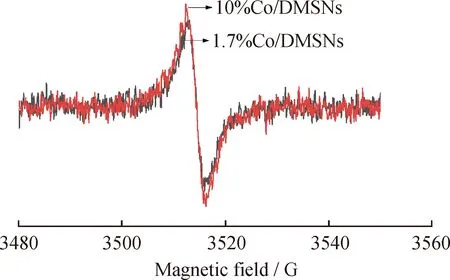
圖9 Co/DMSNs催化劑的EPR圖譜Fig.9 EPR spectra of Co/DMSNs catalysts
2.2 催化劑活性評價
當甲苯體積分數為50 μL/L和空速為15000 mL/(g·h)時,考察了不同Co負載量的Co/DMSNs催化劑上甲苯催化氧化反應性能,結果如圖10所示。由圖10可以看出,所有催化劑的甲苯轉化率隨著反應溫度的升高而增加。轉化率為50%和90%的反應溫度表示為T50和T90,催化劑中Co負載量對T50和T90的影響如圖11所示。由圖11可知,達到相同反應轉化率的反應溫度,隨著Co負載量的升高,總體趨勢表現為先降低后升高。其中Co負載量為10%的催化劑的高溫催化活性最好,當反應溫度為280 ℃時,甲苯幾乎已被完全轉化。在270 ℃時,10%Co/DMSNs催化劑具有最高的轉化率,達到96%左右;在相同的10%Co負載量下,以普通的SiO2為載體的催化劑(10%Co/SiO2)的轉化率明顯較低。
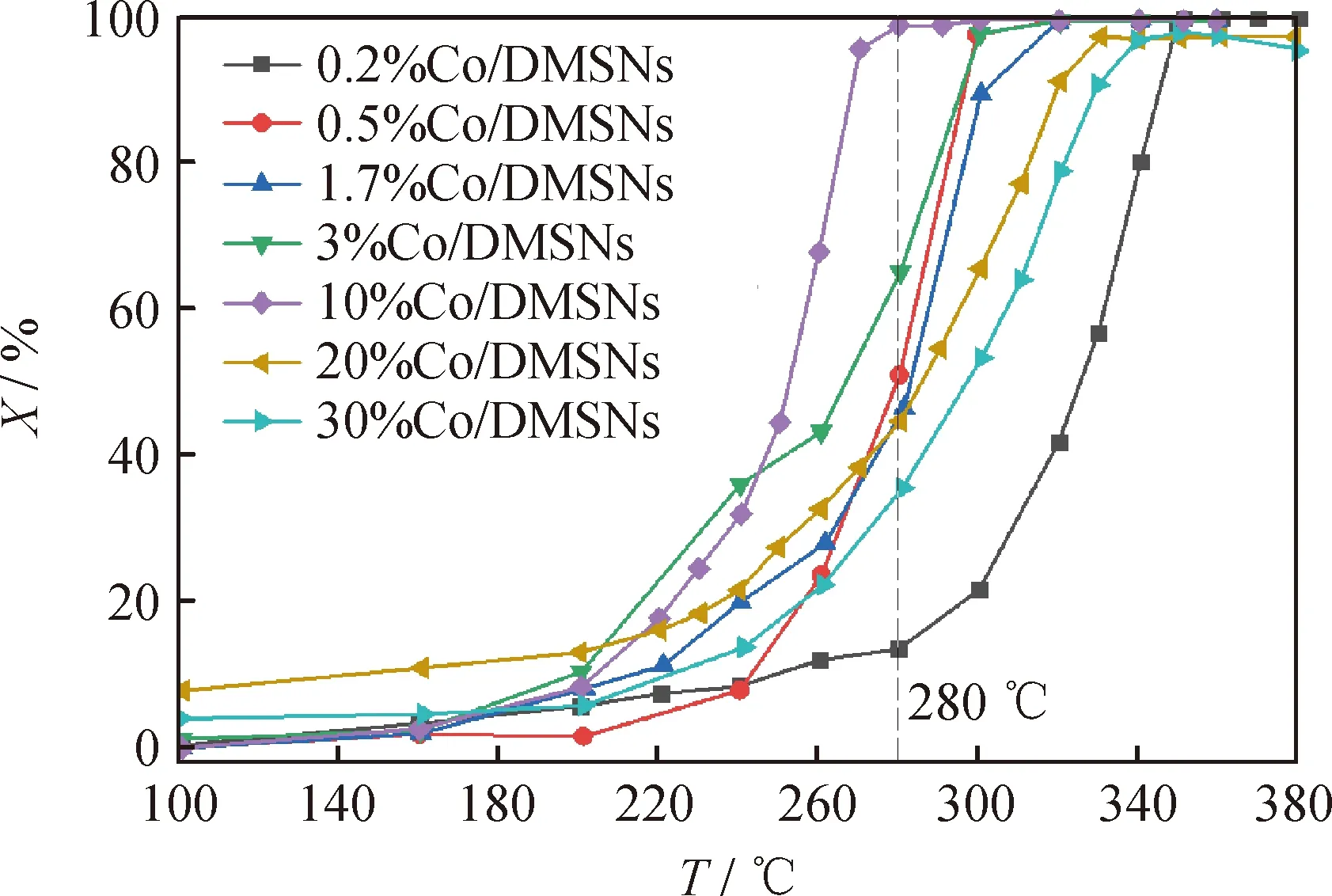
圖10 不同鈷負載量的Co/DMSNs去除甲苯的催化活性Fig.10 Catalytic activity of Co/DMSNs with different cobalt loadings for toluene removalReaction conditions: LHSV=15000 mL/(g·h);φin(Toluene)=50 μL/L
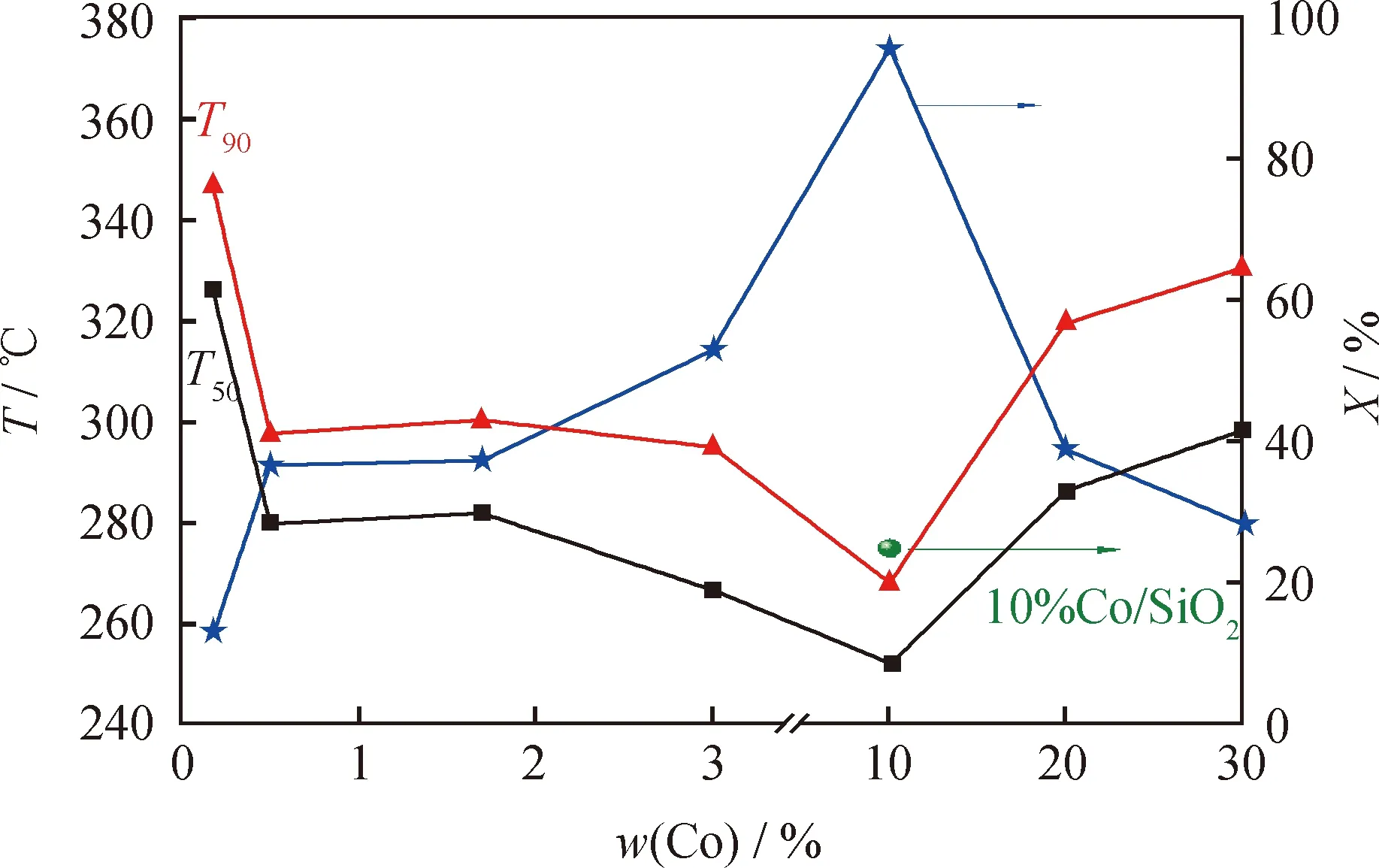
T50 and T90 are the temperatures at 50% and 90% toluene conversion.圖11 Co/DMSNs的T50、T90及270 ℃時的甲苯轉化率隨Co負載量的變化趨勢Fig.11 Change of T50, T90 and the toluene conversion rateat 270 ℃ of Co/DMSNs with Co loadingsReaction conditions: LHSV=15000 mL/(g·h);φin(Toluene)=50 μL/L
結合催化劑表征數據,發現甲苯高溫催化氧化活性最好的催化劑10%Co/DMSNs具有以下特征:(1)催化劑較好地保持了載體的形貌結構,其特殊的球形形貌和中心-徑向孔道結構可以暴露更多的反應位點,也有利于反應物和產物的擴散。(2)活性組分在載體表面的分散度較高,沒有出現大顆粒的晶相氧化物。(3)10%Co/DMSNs表面活性氧物種豐富,表面活性氧物種在氧化反應中起重要作用,這些吸附氧物種能夠促進氧化反應發生[38]。(4)10%Co/DMSNs催化劑表面具有更多的Co2+物種,文獻[7,39-40]報道Co2+物種是Co基催化劑催化氧化VOCs的主要活性位。(5)10%Co/DMSNs具有更多的結構缺陷和氧空位濃度,有利于氧分子在活性中心的吸附活化,從而促進催化氧化反應的進行。而當Co負載量較高時,過量的鈷氧物種造成了載體孔道的堵塞,而且形成明顯的Co3O4顆粒,對其甲苯催化氧化造成不利影響。
進一步研究了甲苯體積分數對10%Co/DMSNs催化劑的甲苯催化氧化性能的影響,結果如圖12所示。圖12結果表明,甲苯體積分數從500 μL/L降低到50 μL/L時,10%Co/DMSNs催化劑去除甲苯的催化氧化活性明顯提高。在甲苯體積分數分別為500、200和50 μL/L時,甲苯的T90分別是353、328和268 ℃。10%Co/DMSNs催化劑在較低的甲苯濃度下展現優異的催化性能,表明催化劑對于低濃度甲苯的脫除具有較好的效果。
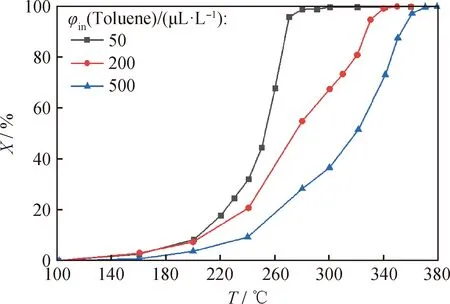
圖12 不同甲苯初始體積分數下10%Co/DMSNs去除甲苯的催化活性Fig.12 Catalytic activity of 10%Co/DMSNs for toluene removal at different initial volume fractions of tolueneReaction condition: LHSV=15000 mL/(g·h)
在實際應用中,催化劑的穩定性及反應氣氛中的水汽等因素都會影響催化劑的性能,因此進一步考察了催化劑的穩定性及抗水性能。在甲苯催化氧化過程中,測定了10%Co/DMSNs催化劑甲苯轉化率隨反應時間的變化,如圖13所示。由圖13(a)可以看出,反應22 h過程中,甲苯轉化率沒有明顯下降趨勢,表現出優異的反應穩定性。在此基礎上,進一步考察了10%Co/DMSNs催化劑在甲苯催化氧化中水蒸氣體積分數對去除甲苯的活性影響。由圖13(b)可知,在反應溫度為260 ℃時,當原料中引入10000 μL/L的水蒸氣后,10%Co/DMSNs催化劑的催化活性出現一定的下降,在通水蒸氣期間,活性基本保持穩定。將水蒸氣關掉后,催化劑的活性基本可以恢復初始水平。因此,10%Co/DMSNs催化劑表現出了一定的抗水性能。對于甲苯的氧化,水蒸氣的活性抑制作用是由于水和甲苯以及氧分子的競爭吸附所致[41]。此外,對反應后的10%Co/DMSNs催化劑進行拉曼表征,結果顯示反應后的催化劑結構未發生明顯的變化而且沒有產生積炭峰。熱重分析結果同樣顯示反應前后的失重曲線幾乎一樣,說明催化劑中未形成明顯的積炭物種,具有良好的穩定性。
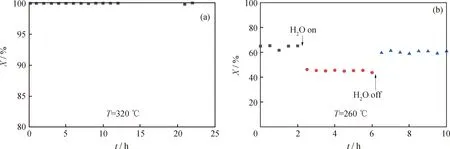
圖13 10%Co/DMSNs催化劑的穩定性及水蒸氣對催化活性的影響Fig.13 Stability of 10%Co/DMSNs catalysts and effect of water vapor on catalytic activity(a) Stability; (b) The effect of water vapor on catalytic activityReaction conditions: LHSV=15000 mL/(g·h); φin(Toluene)=50 μL/L; φin(H2O)=10000 μL/L(Fig(b))
3 結 論
以樹枝狀介孔納米球DMSNs為載體,通過等體積浸漬法制備不同Co負載量的Co/DMSNs催化劑。運用多種表征手段對催化劑進行檢測分析,評價了催化劑催化氧化去除甲苯的反應性能。織構特征分析表明,以DMSNs納米球為載體制備的催化劑具有較高的比表面積和較大的介孔結構,且鈷氧物種分散度較高,這與催化劑獨特的樹枝狀介孔和納米球結構密切相關。這種結構提供的納米空間,抑制了活性組分Co納米顆粒的團聚,促進了其在催化劑表面的分散。負載量影響表面Co2+/Co3+摩爾比,表面Co2+/Co3+摩爾比的增加會提高催化劑的催化活性。活性氧物種是甲苯氧化的關鍵物種,表面Co2+物種是氧活化的主要中心,豐富的Co2+離子有利于氧氣的活化,從而改善其甲苯催化氧化性能。在甲苯催化反應中,10%Co/DMSNs催化劑表現出較高的催化氧化能力,在280 ℃即可將甲苯完全轉化,并表現出良好的穩定性和耐水性。
猜你喜歡
課堂內外·初中版(科學少年)(2025年1期)2025-02-28 00:00:00
課堂內外·初中版(科學少年)(2025年2期)2025-02-28 00:00:00
英語世界(2023年10期)2023-11-17 09:18:18
科學大眾(中學)(2019年3期)2019-05-17 10:04:30
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
汽車觀察(2018年10期)2018-11-06 07:05:26
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
