磷石膏熱分解研究現狀
2022-10-10 11:35:52吳泳霖陳建軍
硅酸鹽通報 2022年9期
吳泳霖,張 偉,奠 波,陳建軍
(1.云南農業大學資源與環境學院,昆明 650201;2.云南師范大學能源與環境科學學院,昆明 650500)
0 引 言
磷石膏是磷化工濕法制取磷酸時產生的工業廢渣[1],反應方程式見式(1)。磷石膏的主要化學成分為CaSO4·2H2O,占總質量的80%以上[2],其余雜質為Ca3(PO4)2、Ca(H2PO4)2·H2O、CaHPO4·2H2O、殘酸、氟化物、重金屬、有機物和少量放射性元素[3]。每生產1 t磷酸約產生5 t磷石膏副產物[4]。據不完全統計[5-6],全球磷石膏總堆放量已超過60億t,且每年的產量高達1億t。目前,我國磷石膏儲存量已超過2.5億t[7],且每年產量保持在7 500萬~7 800萬t,但資源化利用率僅為40.0%[8]。磷石膏的堆存不僅占用大量土地資源,且其中的雜質會隨地表徑流進入土壤以及地下水中造成嚴重污染[9-10]。因此,為實現環境保護和綠色可持續發展,磷石膏的資源化利用迫在眉睫。
目前,國內外磷石膏資源化綜合利用的途徑主要為生產建筑材料,如半水石膏材料[11]、建筑石膏或石膏板[12]、制硫酸聯產水泥[13]等。在生產上述建筑材料的過程中,磷石膏的熱處理是關鍵環節,將直接影響產品的能耗和質量。在不添加還原劑和催化劑的條件下,磷石膏的熱分解存在能耗高、成本高和分解率低等問題[14],反應方程式見式(2)。目前,針對磷石膏直接熱分解技術的缺點,國內外學者的研究方向逐漸轉移到篩選熱分解時的還原氣氛[15]、還原劑[16]和催化劑[17]方面,以降低分解溫度,提高分解率。本文綜述了近年來關于磷石膏熱分解的研究進展,為今后磷石膏熱分解技術的發展提供參考。

(1)
(2)
1 還原條件下磷石膏的熱分解
磷石膏在N2或空氣氣氛下熱分解,不僅分解率低,且反應起始溫度高。此外,研究[18]表明空氣中的O2會使CaSO4的熱穩定性增加,抑制磷石膏的分解,因此,采用添加還原劑的方法促進磷石膏熱分解成為當下的研究熱點。
1.1 CO氣氛條件
大量研究試驗證明CO作為磷石膏熱分解的還原氣氛,能有效降低分解初始溫度,提高分解率和分解速率。在CO氣氛下磷石膏熱分解反應過程中會發生如下反應:
(3)
(4)
(5)
在CO氣氛下,磷石膏分解后的固體產物主要是CaO和CaS,氣體產物是CO2和SO2,反應機理[19]為CO中的C—O鍵和CaSO4中的S—O鍵被激活和拉伸,導致CaSO4中的O原于被逐漸剝離,得到最終產物CaS。反應流程見式(6)。
(6)
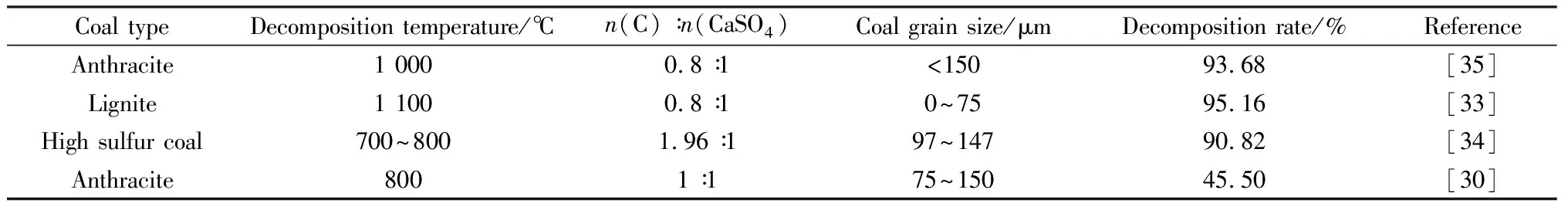
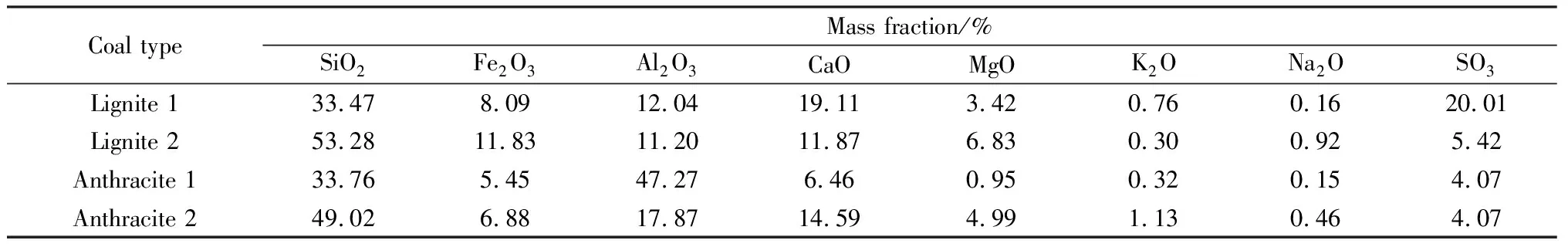
氣固反應(3)和(4)分別是吸熱反應和放熱反應, 在低濃度CO氣氛下以反應(3)為主,反之則以反應(4)為主[20]。隨著溫度逐漸升高,反應(3)和反應(5)的占比增加[21],CaSO4大量分解,產物中的CaS繼續和CaSO4發生固-固反應生成CaO,并釋放SO2[22]。Zheng等[23]根據熱力學計算結果,結合不同溫度下磷石膏分解產物的X射線衍射譜和X射線熒光光譜分析發現,CaS和CaSO4的特征峰隨著反應溫度的升高而降低,在1 100 ℃下,CaS的特征峰幾乎消失,CaO含量達到最大值。同時,CO含量的增加有利于磷石膏在低溫下的分解,溫度越高,固體產物中CaS越少,CaO越多。Ning等[24]研究了磷石膏在純CO、10%(體積分數)CO和N2氣氛中的熱分解及動力學過程,通過XRD譜分析分解后的殘渣發現:在純CO氣氛中,磷石膏分解的主要產物為CaS;在N2氣氛中,磷石膏分解產物主要為CaO并伴隨有少量CaS;在10%CO氣氛中,磷石膏分解產物同時存在CaO和CaS;在三種氣氛下,磷石膏分解活化能大小為純CO<10%CO CO已被證明能有效促進磷石膏熱分解,但CO是生產有機化工的重要原料,將其作為磷石膏分解的還原氣氛會給工業生產帶來成本壓力。因此,尋找更廉價和易得的氣氛代替CO成為新的研究目標。有研究者采用廢氣中的H2S作為磷石膏還原氣氛,其反應見式(7)。H2S氣氛下磷石膏分解分三個階段,當溫度在600~950 ℃時磷石膏與H2S反應生成CaS;當溫度達到1 000~1 150 ℃時反應(5)和(7)形成平行競爭,主要分解產物為CaS和CaO;繼續升溫,主要分解產物從CaS向CaO轉換[26]。此外,在一定程度內隨著H2S流量增加,磷石膏的分解率顯著提高[27]。不同氣氛下磷石膏的分解條件和相關產物見表1。 (7) 通過對比幾種氣氛發現,在空氣和N2氣氛下磷石膏主要產物為CaO,N2對磷石膏的分解無明顯影響。空氣中的O2會在一定程度內抑制磷石膏分解,但混合CO時可以提高目標產物CaO的產率。與其他氣氛下相比,CO降低磷石膏分解溫度、提升分解率的效果最為顯著,但會降低目標產物CaO的產率。此外,CO作為合成氣的主要原料,用于磷石膏的熱分解時需要考慮經濟效益。將廢氣中的H2S作為磷石膏還原氣氛,能有效提高其分解率,降低分解溫度,達到廢物利用的目的,具有較高的環保意義和經濟價值,但目前該研究深度不夠,機理尚不明確,工藝尚不成熟,無法應用到實際生產中。 早在1915年,Muller和Kuhner提出在高溫條件下以焦炭為還原劑分解磷石膏的方法,并于1916年經過工廠試驗將其應用到實際生產中并沿用至今[28]。炭還原條件下磷石膏熱分解主要發生高溫固相反應,見式(5)、(8)、(9)。此外,研究[29]表明,與N2和空氣氣氛下相比,在添加焦炭的條件下,磷石膏的分解溫度顯著降低。 (8) (9) 煤炭的種類、摻炭量與摻炭粒徑是影響磷石膏分解的主要因素。表2為不同種類煤炭作為還原劑時磷石膏的分解率。 表2 不同種類煤炭作還原劑時磷石膏的分解率 摻入煤炭的種類不同,對磷石膏分解的影響也不同,這是由于不同煤炭的礦物含量有所差異(見表3),在沒有礦物的條件下,C與CaSO4幾乎不反應[30]。煤在形成過程中煤化程度不同,在壓力和溫度的作用下從煤化程度較低的低階煤逐漸演變為煤化程度較高的高階煤,煤種按煤化程度從低到高排列為:褐煤、亞煙煤、煙煤、無煙煤,且低階煤比高階煤的熱穩定性更低[31],更易發生反應。此外,煤炭中的Fe2O3能有效促進磷石膏分解,褐煤中Fe2O3的含量普遍高于煙煤,因此相較于高階煤(煙煤),礦物含量更高、熱穩定性更低的低階煤(褐煤)更適用于磷石膏分解。 表3 不同種類煤炭所含礦物及含量[30] 對于摻炭量而言,隨著炭摻入量的增多,磷石膏分解初始溫度會略有降低[32],但摻炭量并非越多越好,Zheng等[33]研究發現炭與磷石膏的最佳摩爾比為0.8∶1,當摻炭量繼續增加時,磷石膏分解受到抑制。 煤炭粒徑也是影響磷石膏分解的主要因素,粒徑越小越易與磷石膏混勻,使分解反應更充分;相反,粒徑過大的炭源由于接觸面小不利于反應進行。但選取摻入炭源粒徑會有一定的下限,這是由于當煤炭粒徑過小會造成一部分煤炭發生氧化反應產生CO,造成煤炭的損失,從而不利于磷石膏分解[34]。 我國磷石膏分解的焦化還原工藝從20世紀50年代開始沿用至今,工藝成熟,但仍存在資金投入大、能耗高、煙氣中SO2濃度低、CO2排放量大、操作彈性小等缺點[36]。因此研究者們提出了以硫代替碳作還原劑分解磷石膏,反應如式(10)所示。 (10) 由于硫磺還原條件下磷石膏的分解以高溫固相反應(5)、(10)為主,因此氣相產物只有SO2,升溫過程中,硫磺經歷了從固相到液相再到氣相的形態轉變[37]。650 ℃時以反應(10)為主導,1 100 ℃以后以反應(5)為主導[38],升溫過程中達到熱力學平衡時各物質的組成見圖1。由圖1可知:300 ℃以內硫主要以固相的形式存在;到達400 ℃時固相硫消失,體系中的硫主要以氣相S8存在,隨著溫度升高,硫的形態從八個原子變成兩個原子;當溫度上升到700 ℃時,硫主要以氣相S2形態存在;當升溫至750 ℃以后,磷石膏與S2開始反應,硫含量逐漸降低,SO2和CaS含量逐漸升高。Yang等[39]通過計算對比發現,硫還原分解過程總反應焓(236.979 kJ/mol)小于焦炭還原分解過程總反應焓(306.245 kJ/mol)。理論上表明,與焦炭相比,硫磺作為還原劑分解磷石膏的能耗更低。 圖1 硫磺熱分解磷石膏過程元素轉化[38] 焦炭作為我國現階段主要的磷石膏還原劑,效果顯著,工藝成熟,但存在成本高、CO2排放大等問題,為響應國家“碳達峰,碳中和”政策,急需尋找更環保的材料代替焦炭。有研究者通過模擬計算和實驗發現,與傳統煤炭相比,以硫磺作為還原劑分解磷石膏,能耗更低,目標產物SO2的濃度更高,具有更高的應用價值,但現階段該方向研究不足,工藝不成熟,因此對該工藝實際應用的完善尤為重要。 由于磷石膏需在較高溫度下才能分解,且分解率和目標產物含量低,而添加合適的催化劑可以有效降低分解溫度,提高分解率,增加目標產物含量。催化劑主要分為Ca[40]、Fe[41-42]、Al[43]和Si[44]基催化劑。 當前針對Ca基催化劑的研究較少,僅存在關于CaF2催化劑的相關報道,其催化機理是在溫度較低時,CaF2與CaSO4·2H2O反應形成中間過渡相氟硅酸鹽,促進磷石膏的分解;當溫度大于900 ℃時,CaF2與磷石膏生成的氟、硫共融物也會促進分解過程中的固相反應,顯著降低磷石膏分解溫度[45]。Feng等[46]研究發現:CaF2促進了Ca2SiO4和氟硅酸鹽絡合物(3CaO·2SiO2·CaF2)的形成,促使磷石膏在低溫和弱還原氣氛下分解;對比無催化劑條件下,添加一定量的CaF2能有效提高磷石膏的分解率和SO2的轉化率。 目前針對Fe基催化劑的研究較為深入,主要有FeCl3和Fe2O3及其鐵氧化物催化劑。在磷石膏與FeCl3浸漬過程中,磷石膏與FeCl3發生離子交換反應,見式(11)。在CO還原氣氛條件下,隨著溫度升高會產生FeS、FeO、Fe3O4和Fe2O3等一系列含鐵化合物作為反應介質[47],見式(12)~(15)。Fe2O3催化劑加速了CaSO4在還原反應過程中中間產物CaFeSO的形成,隨著反應進行,CaFeSO含量逐漸減少,最終形成CaS[48],如式(16)~(18)所示。Zheng等[49]通過熱重分析發現,相較于磷石膏原樣,浸漬過FeCl3后磷石膏分解初始溫度降低了300 ℃,質量損失增加14.3%,證明Fe及其氧化物能夠極大地促進磷石膏分解,顯著降低磷石膏分解初始溫度。在常溫下,與CaO相比,CaCl2的標準摩爾生成焓更小,因此更易生成促進磷石膏分解的物質,降低分解溫度以及提高分解速率。 (11) (12) (13) (14) (15) (16) (17) (18) 目前關于Al基催化劑的相關報道僅有Al2O3,其作用機理是在CaSO4分解初期,Al2O3與CaSO4反應形成鋁酸鈣(CaAl2O4),使CaSO4的熱穩定性和分解溫度降低,加速CaSO4的分解,見式(19)。隨著反應進行,中間產物CaAl2O4與CaSO4進一步反應形成硫鋁酸鈣(CaO·3Al2O3·CaSO4),見式(20),這阻礙了CaSO4的進一步分解[50]。Yang等[51]通過FactSage計算分析了CaSO4在C-Al2O3體系下的熱分解過程,推測出可能發生的反應如式(21)~(25)所示,Al2O3的加入使磷石膏分解初始溫度降低。磷石膏分解產物中含有硫鋁酸鈣(Ca4Al6O12(SO4)),改變了CaSO4的分解路線。 (19) (20) (21) (22) (23) (24) (25) 針對Si基催化劑的研究,現階段主要發現的是SiO2和一些硅酸鹽礦物。SiO2對磷石膏分解的影響顯著,其作用機理[50]是SiO2與CaSO4在高溫下發生固相反應,形成Ca2SiO4和CaSiO3,見式(26)。該反應促使CaSO4不斷暴露出新的接觸面,使其性質更加不穩定,從而導致磷石膏分解速率和SO2的釋放速率提高;隨著反應的進行,固相產物Ca2SiO4與CaSO4進一步反應,形成一種會抑制磷石膏分解的中間產物硫硅酸鈣(CaO·2SiO2·CaSO4),見式(27);隨著溫度繼續升高,CaO·2SiO2·CaSO4再次分解為Ca2SiO4和CaSO4,CaSO4的分解速率回升。Shi等[52]研究發現磷石膏在熱分解過程中隨著SiO2含量增加逐漸生成一系列硅酸鹽礦物,這些硅酸鹽礦物的熔點是可變的,因此反應的熱分解溫度降低。此外,添加硅酸鹽礦物也能在一定程度上降低磷石膏分解初始溫度,提高其分解速率,如鉀長石(KAlSi3O8)在熱分解過程產生中間產物SiO2,促進了磷石膏分解[53-54],高溫固相反應見式(28)、(29)。 (26) (27) (28) (29) 在各種催化劑中,CaF2催化劑能有效降低磷石膏的分解溫度,提高SO2的轉化率,但目前研究較少,機理不明確,工藝不成熟,還不能應用到實際生產中。中國作為世界上最大的鋁生產國[55],具有豐富的Al2O3資源,同時我國的SiO2資源也極為豐富[56],以Al2O3和SiO2作為催化劑具有來源豐富、成本較低的優勢。Al2O3和SiO2作為磷石膏分解的催化劑,在分解初期可加快反應,降低初始溫度,但在反應過程中會產生硫硅酸鈣和硫鋁酸鈣等中間產物,抑制磷石膏分解,因此Si和Al基催化劑作為磷石膏分解催化劑并不完美。當前Fe基催化劑在磷石膏熱分解中應用最廣泛、研究最深入、效果最顯著。但從經濟效益的角度考慮,需要尋找更具經濟性的新型Fe基催化劑,進一步降低成本。 熱分解是磷石膏資源利用的關鍵,當下磷石膏熱分解研究的重點主要為兩方面:一方面是尋找成本更低、來源更豐富的新型還原劑和催化劑;另一方面是將現有氣氛、還原劑和催化劑進行協同聯用,以達到更高的分解率和目標產物含量,以及降低能耗的目的。現對近年來的研究提出以下建議: (1)在還原劑、催化劑和還原氣氛的類型上可以嘗試更多選擇,使用還原性廢氣代替工業原料CO,使用更加環保、更具經濟適用的生物炭代替煤炭,使用來源廣泛、無需加工的天然礦物代替需要制備的傳統催化劑,以達到資源利用最大化的目的。 (2)近年來有研究者嘗試選擇更多還原劑和催化劑,具有一定的研究價值,但現階段研究深度較淺,機理尚不明確,一部分結果來源于模擬計算,缺乏實驗數據支撐,在未來可加深研究,明確機理。 (3)單一的氣氛、還原劑和催化劑難以達到在降低分解溫度的同時提高分解率和保證目標產物的產率,缺乏繼續研究價值,而未來的研究將集中在多氣氛、還原劑和催化劑的搭配,以及各組分之間的配比,明確機理。 (4)多氣氛、還原劑和催化劑的聯用相較于單一使用能夠提高分解率和目標產物產率,但也提高了材料成本和工藝流程復雜程度,因此在今后研究中需增加此類問題的考慮。1.2 H2S氣氛條件
1.3 焦炭條件
1.4 硫磺條件

2 催化條件下磷石膏的熱分解
2.1 Ca基催化劑
2.2 Fe基催化劑
2.3 Al基催化劑
2.4 Si基催化劑
3 結語與展望
猜你喜歡
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
汽車工程學報(2017年2期)2017-07-05 08:13:02
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
