Dual RNA-seq技術及其在宿主-病原體相互作用中的研究進展
2022-10-25 00:53:46夏應菊趙俊杰劉業兵宋振輝張乾義
中國獸藥雜志 2022年9期
鄒 宏,夏應菊,徐 璐,趙俊杰,李 玲,王 震,劉業兵,王 琴,宋振輝,張乾義*
(1.西南大學動物醫學院,重慶榮昌 402460;2.中國獸醫藥品監察所,北京100081)
轉錄組是指特定組織或細胞在某一功能狀態下轉錄出來的所有RNA的總和,包括mRNA(2%~4%)、tRNA(5%~15%)、rRNA(80%~90%)和ncRNA(1%)等[1]。RNA-seq是21世紀初進行轉錄組分析的新一代測序技術,利用RNA-seq可以全面快速的獲得組織和細胞在某一狀態下幾乎所有的轉錄本序列和表達信息,進而研究不同發育階段或組織之間的基因差異表達模式,同時還可發現新的基因和轉錄本,為未知蛋白的功能研究提供基因序列參考信息[2]。隨著RNA-seq數據的累積,科學家們建立了廣泛的轉錄組數據庫[3]。伴隨高通量測序深度不斷提高,2012年Westermann等[4]在RNA-seq的基礎上提出并探討了Dual RNA-seq技術的可行性,Dual RNA-seq得以實現并應用到生命科學領域的研究中[5]。與RNA-seq相比,Dual RNA-seq可以同時監測宿主和病原體的所有編碼和非編碼轉錄本[6],無需分離病原體和宿主、避免分離樣本造成的干擾和數據浪費,揭示兩個轉錄本之間基因表達的動態變化,同時通過相互作用模型圖預測基因的調控關系和兩物種間的相互作用機制,為進一步研究病原體致病機制提供重要依據。
1 Dual RNA-seq概述
Dual RNA-seq技術的發展和應用與基因測序技術的發展密切相關。基因測序技術的發展按照時間段可分為第一代毛細管測序、第二代高通量測序和第三代單分子測序技術。第一代毛細管測序以1977年Sanger建立的“DNA雙脫氧鏈末端終止測序法”為基礎,但Sanger測序法的通量很低,不能實現大批量測序,而轉錄組中有成千上萬個序列,因此Sanger法難以應用在轉錄組測序的研究中[7]。高通量測序技術,即二代測序(Next generation sequencing,NGS)技術,其測序平臺主要為454測序、Solexa及SOLID測序等[1]。與一代相比,二代測序的主要特點是以高密度基因芯片的熒光成像為基礎,測序通量大幅度提升,測序時間和成本顯著下降[8],可以一次對幾十萬到幾百萬條DNA分子序列測定,使物種全轉錄組和基因組的整體分析成為可能[9],實現了測序的自動化和高通量,促進了轉錄組學研究的快速發展。三代測序技術,又叫單分子測序技術[10](Single Molecule Real-Time sequencing,SMRT),通過檢測標記熒光獲得序列信息的HeliScope遺傳分析系統和SMRT技術,實現了對每一條DNA分子單獨測序,其讀數較二代更長(最長可高達60 kb),具有無GC偏好性,無聚合酶鏈式反應偏向性擴增等特點。
Dual RNA-seq是在NGS技術的基礎上發展起來的,其核心除了高通量熒光成像系統,此外還包括高密度生物芯片、高通量圖像傳感器及高功率半導體激光器。Dual RNA-seq測序深度決定了技術的準確性和可行性[13]。轉錄組測序會受到研究樣本遺傳材料的復雜性和大小的限制,如真核生物感染宿主時往往涉及兩種完全不同的互作生物體轉錄組,通常轉錄組測序技術更加側重于分析參與蛋白翻譯的mRNA,忽略了大量非編碼區RNA[14],而非編碼RNA廣泛參與生命過程的各個環節,如生長、發育、分化、免疫,甚至在腫瘤的形成中也具有重要的調節功能,蛋白質和DNA發揮正常的生理學功能離不開非編碼區RNA[15]。隨著高通量(High-Throughput sequencing,HT)測序技術深度的不斷提升,宿主轉錄組和病原體轉錄組同時測序技術得以實現,Dual RNA-seq技術的應用更加廣泛[15],進一步加強了對病原體感染宿主期間發生的分子相互作用(包括非模式宿主物種)的分析。Dual RNA-seq技術改進了文庫構建和測序技術,可以深入研究信號通路相關的非編碼RNA,及研究感染相關的sRNA、線粒體RNA等[14],使其可以同時捕捉病原體和宿主中所有類別的編碼和非編碼RNA,使我們更好地了解感染過程中病原體和宿主的生理變化,揭示了在RNA-seq中不可見的毒力相關小非編碼RNA隱藏的分子表型。
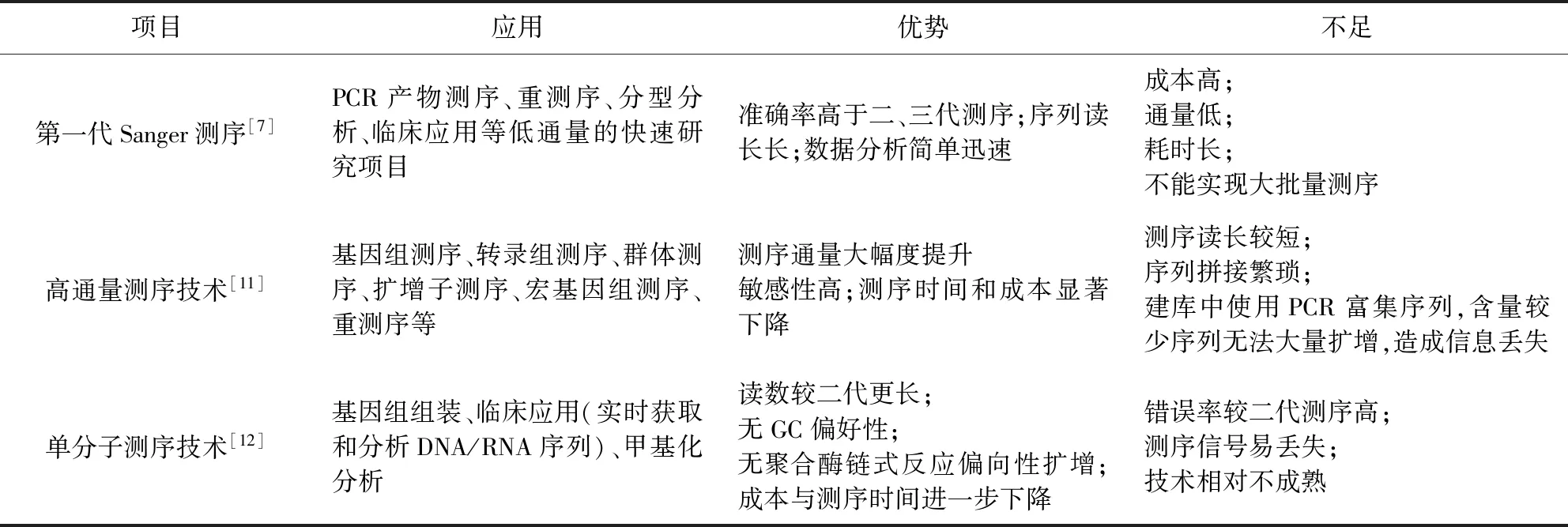
表1 基因測序技術發展特征比較Tab 1 Comparison of the development characteristics of gene sequencing technology
Dual RNA-seq的過程與RNA-seq相同,包括轉錄組RNA的提取、cDNA文庫的構建、高通量測序及數據預處理、比對混合基因組和轉錄組整體的質量評估等。其區別在于,在同時分析病原體與宿主轉錄組時,RNA-seq 技術需要分別構建病原體及宿主的cDNA文庫,再將其各自映射到病原體及宿主參考的基因組中,而Dual RNA-seq 技術無需分離兩物種,只構建一個cDNA文庫,然后將測序得到的原始數據經過質量控制和修剪,各自映射到宿主及病原體參考基因組,確定每個基因的讀段數,再利用相關分子生物學技術對數據進行分析[16]。如Dual RNA-seq可以通過構建數字基因表達譜(Digital Gene Expression Profiling,DGE)確定基因差異表達情況。利用GO(Gene Ontology,GO)和KEGG富集(Kyoto Encyclopedia of Genes and Genomes,KEGG)進行層次聚類分析[17],一般這些相同分組的基因具有相似功能并聚于一簇,屬于同一調控途徑。采用 Cytoscape軟件對差異蛋白的 mRNA 建立節點及網絡形式的生物分子相互作用網絡[18],挖掘互作網絡中的核心模塊及關鍵節點,從分子調控網絡的角度出發探究其分子致病機制和發現新藥靶點。
2 利用Dual RNA-seq研究病原體與宿主相互作用的進展
2.1 Dual RNA-seq技術在原核生物研究中的應用
病原體入侵宿主時是一個主動、動態的過程,在病原體感染宿主細胞的過程中,病原體和宿主分別通過入侵策略和自身防御影響另一個生物體,在這種互相影響下的基因表達會發生差異并出現一系列級聯變化反應[4]。利用Dual RNA-seq研究杜克雷嗜血桿菌(Hemophilusducreyi)感染病例[19]時發現,杜克雷嗜血桿菌通過利用抗壞血酸作為一種替代碳源在膿腫中生存,還通過上調參與無機離子、厭氧代謝和營養運輸的基因來適應膿腫,該研究第一次描述了細菌和人類宿主在感染部位的相互作用網絡,揭開了這一系列級聯反應的面紗。
2017年Robert等[20]首次將Dual RNA-seq應用到體內感染研究中,已有文章證實A/J小鼠對金黃色葡萄球菌(Staphylococcusaureus)非常敏感,在C57BL/6具有較強的抵抗性的前提下,研究人員使用金黃色葡萄球菌感染A/J和C57BL/6。利用Dual RNA-seq技術對兩組小鼠轉錄組的高通量測序數據分析,結果表明兩組小鼠感染反應中,大量的宿主基因表達上調,特別是編碼炎癥細胞因子的基因,如白細胞介素-6(IL-6)基因、白細胞介素-1α(IL-1α)基因、白細胞介素-1β(IL-1β)基因和腫瘤壞死因子-α(TNF-α)基因以及參與單核細胞/巨噬細胞螯合作用的趨化因子,如趨化因子(CXC基序)配體1(Cxcl1)、趨化因子(CXC 基序)配體2(Cxcl2)和趨化因子(CXC 基序)配體3(Cxcl3),發現A/J小鼠和C57BL/6小鼠之間的感染組織的微環境是高度不同的,這可能會極大的影響金黃色葡萄球菌的毒力決定因素的表達。另外還發現編碼急性期蛋白的宿主基因,如血清淀粉樣蛋白A1(SAA1)、血清淀粉樣蛋白A2(SAA2)、結合珠蛋白、S100鈣結合蛋白A8(S100A8)和S100鈣結合蛋白A9(S100A9)在感染的A/J和C57BL/6小鼠中被高度誘導。雖然轉錄組整體水平分析結果顯示炎癥相關基因在兩組小鼠對金黃色葡萄球菌感染的反應中都高表達,但A/J明顯高于C57BL/6小鼠。此外,編碼精氨酸酶1(Arg1)和精氨酸酶2(Arg2)的基因在感染A/J小鼠中的表達程度要高于感染C57BL/6小鼠。精氨酸酶可以通過消耗細胞外的L-精氨酸和一氧化氮(NO)的生物利用率,導致內皮的一氧化氮合成酶失調,從而產生高水平的有害活性氧(ROS),從而促進內皮細胞功能障礙。以上研究表明,A/J小鼠和C57BL/6小鼠之間的感染組織的微環境是高度不同的,這可能會極大的影響金黃色葡萄球菌的毒力決定因素的表達。
利用Dual RNA-seq研究肺炎鏈球菌(Streptococcuspneumoniae)感染人體轉錄組學[15,21]發現,人類肺上皮細胞中谷胱甘肽依賴的活性氧解毒途徑是由肺炎鏈球菌產生的活性氧引起的,感染期間添加抗氧化劑白藜蘆醇可抑制這種反應。在肺炎球菌與肺上皮細胞體外感染模型中,肺炎球菌通過對上皮細胞的粘附激活自身糖轉運蛋白,同時抑制宿主趨化因子IL-8的表達和抗菌肽的產生,避免其激活宿主炎癥反應,保護自身不被抗菌肽清除。利用Dual RNA-seq實現了在同一實驗中對宿主和病原體同時監測,從而對肺炎球菌與肺上皮細胞相互作用關系有更清晰的認知。
以上研究證明,通過運用Dual RNA-seq技術使我們能夠系統性的研究病原體感染宿主后如何通過調節差異基因的表達得以探究病原體如何在宿主體內進行吸附、侵入、脫殼、生物合成、組裝和釋放等病毒的復制過程[22],從而能夠研究同一病原體在入侵不同宿主或同一宿主的不同部位時宿主與病原體轉錄組水平的應答特征,了解導致病毒嗜性變化和致病性差異的機制,鑒別致病力相關的關鍵基因,為深入了解復雜的宿主-病原體相互作用網絡和疾病的發生發展提供了有力的支持。
2.2 利用Dual RNA-seq 在真核生物中的研究應用 相較于原核生物,病原體與真核生物相互作用的過程中,病原體感染宿主存在著廣泛的特殊相互作用,如共生、寄生、競爭和拮抗等[23]。根結線蟲(Meloidogyne)是危害亞洲和拉丁美洲水稻主要的病原之一,寄生感染過程中通過分泌蛋白因子來抑制水稻防御和紊亂代謝[24]。在沒有參考基因組的情況下利用Dual RNA-seq技術,Petitot等[20]獲得了66396個根結線蟲和水稻相互作用的轉錄本,進一步研究分析共發現15個可能的關鍵效應基因,其中包括兩個特征明顯的線蟲效應基因CLE和VAP1,以及一個水稻效應基因金屬硫蛋白(Metalthionein)基因,為根結線蟲感染水稻時的生物學特性的研究提供了重要依據。
利用Dual RNA-seq研究核果鏈核盤菌(Ascomycetes)感染不同發育階段的油桃互作轉錄本[25]時發現,宿主防御機制及病原體的感染策略與果實發育階段有密切關系,核果鏈核盤菌只對成熟果實造成病變,在未成熟的果實中,病原體數量,宿主反應及病原體的轉錄活動在感染后14~24 h逐漸增強,此后核果鏈核盤菌不能利用碳水化合物活性酶(CAZymes)進行滲透,而宿主能夠通過調節激素反應和氧化爆發來對抗核果鏈核盤菌的增殖。但在成熟果實中,核果鏈核盤菌更加依賴蛋白質水解效應,更利于感染早期核果鏈核盤菌絲狀體的生長。對感染核果鏈核盤菌的成熟果實激素分析表明,雖然茉莉酸活性可能有利于防御,但成熟果實釋放的高乙烯活性可能通過誘導成熟過程促進核果鏈核盤菌易感性。最后通過進一步驗證,確定了在不同生長發育階段感染中顯著上調的核果鏈核盤菌基因,這些基因可能成為控制褐腐病的靶點。
此外,Musungu等[26]用黃曲霉(Aspergillusflavus)感染玉米3 d后,利用Dual RNA-seq構建了基因共表達網絡(Gene Co-expression Network Analysis,GEN),分析發現GEN證明了茉莉酸,活性氧和乙烯等許多已知途徑具有高度連通性,確定了泡囊與產生黃曲霉毒素之間的互作網絡,發現了一個重要的黃曲霉毒素簇調節因子AflS與互作網絡中的多個參與生存活性氧的基因共同調控,表明AflS可以監視宿主活性氧水平。此研究發現玉米和黃曲霉的整個基因共表達網絡及種間相關子集,該技術可作為發現黃曲霉侵染玉米的早期診斷工具。
2.3 利用 Dual RNA-seq在病毒中的研究應用 目前在病毒轉錄組學的研究中還是主要以RNA-seq技術為主[27],如利用RNA-seq技術對高致病性禽流感H5N1病毒轉錄組高通量測序分析中發現,H5N1通過下調IFNAR1基因和IFNAR2基因來減弱干擾素誘導信號,從而實現逃避宿主先天免疫的目的[28]。豬繁殖和呼吸障礙綜合癥病毒(Porcine reproductive and respiratory syndrome,PRRSV)感染豬的研究過程中發現,PRRSV會導致宿主體內的IRF7以及其他一些誘導抗病毒的信號因子基因(Interferon stimulated gene,ISG)下調,通過構建基因表達譜分析發現,PRRSV病毒非結構蛋白nsp7基因上調是導致這些抗病毒細胞因子下調的主要原因[29]。在豬瘟病毒(Classical Swine Fever Virus,CSFV)強毒株感染豬的動物模型[30]上,發現豬體內細胞凋亡相關基因、免疫應答相關基因以及新陳代謝相關基因均有不同程度的差異表達,揭示CSFV可能通過抑制或改變這些基因的表達來實現潛伏、復制和傳播。以上研究結果均是在RNA-seq的基礎上針對動物病毒或其宿主進行的單方面轉錄組學分析,缺乏另一方面的研究數據及辯證關系的分析佐證。然而隨著高通量測序的快速發展,Dual RNA-seq技術的出現使得在宿主轉錄組中同時分析病毒RNA基因組和發現病毒感染生物標志物成為可能,隨之被逐漸應用到病毒與宿主相互作用的研究中[31]。該技術可以對宿主和病原體的RNA進行同時捕獲和分析,能夠篩選基因差異表達譜的幾乎所有差異表達的基因,為宿主與病原體的相互作用機制的研究提供強有力的依據,該技術在病毒轉錄組學研究中引起人們極大的興趣。Giulia等用H3N2流行毒株Brisbane/10/07和Perth/16/09以及早期毒株Udorn/307/72分別感染人的支氣管上皮細胞BEAS-2B[33],利用Dual RNA-seq對病原體和宿主轉錄組數據進行分析時發現,以上三個毒株分別有81、372和1614個基因發生差異表達,這三個毒株可引起22個相同宿主基因表達上調,包括具有抗病毒作用的ISG宿主基因[32](如MX1、RO、RSAD2和Ieft1-3)、誘導IFN表達基因(DDX58和DDX60)、抑制IFN表達的基因(泛素類ISG15和E3連接酶IMTR69),或減弱IFN信號傳導基因(USP18和GBP4,后者抑制IRF7),而在三株不同的H3N2毒株感染宿主后IFNAR1基因均發生了下調[34],且差異表達基因(Differentially Expressed Genes,DEGs)分析發現下調程度并沒有明顯差異,但通過Dual RNA-seq技術卻發現三株不同病毒株在宿主感染過程中引起了一系列相同的級聯反應,具體的反應途徑還需進行下一步研究。文章揭示了H3N2與人上皮細胞相互作用的新要素,并強調了Dual RNA-seq在確定基因組水平的分子變化方面的重要性。
此外,Dual RNA-seq技術也可以作為一種診斷方法,Wesolowska等采集了92例哮喘兒童及69例健康兒童臨床樣本,使用qPCR對六種常見的呼吸道病毒進行篩查。結果顯示21例病毒陽性的患者中有19例為哮喘患者,2例為健康兒童。將這兩例篩選結果為陰性的哮喘兒童樣本進一步研究,使用CCL8和CXCL11(這兩種被確定為急性呼吸道疾病上調最明顯的病毒生物標志物基因)進行qPCR檢測,發現這兩例患者的樣本中這兩種生物標志物顯著表達。進一步對這些樣本進行Dual RNA Seq分析發現在這21個qPCR陽性樣本中有18個樣本檢測到呼吸道病毒,有15個樣本通過Dual RNA Seq檢測到的呼吸道病毒與qPCR鑒定的病毒相符,有3個被qPCR陽性的樣本測序發現實際為人類腸道病毒及人類冠狀病毒。該文章證實利用Dual RNA-seq在診斷上比qPCR更準確更敏感,為采取正確措施控制疾病發展發揮更精準的作用,體現了宿主和病原體的Dual RNA Seq是一種可以檢測病原體及揭示其對宿主轉錄組影響的開創性方法。與常規的qPCR檢測及傳統方法(免疫層析、直接熒光抗體等技術等)相比,Dual RNA Seq是基于比qPCR結果更全的病毒基因組序列檢測,具有更高的特異性、不受引物交叉反應的限制還可以根據少量的讀數對病毒進行分型,而qPCR方法卻難以實現[35-36]。Dual RNA Seq 可以從樣本中檢測病毒并確定其對宿主細胞轉錄組的影響,對于了解由病毒感染及后遺癥引發的復雜疾病至關重要,在臨床疾病診斷中意義重大。
3 總結和展望
本文就Dual RNA-seq技術本身及在病原體與宿主相互作用的研究中的應用進行論述,同時也介紹了它在生命科學領域研究中的優勢,但是Dual RNA-seq技術也有一定的局限性,例如要求有較高RNA濃度,由于細菌和真菌的細胞壁厚,破壞時要比哺乳動物細胞處理更困難,處理過程會導致哺乳動物細胞RNA濃度低,影響后期數據的分析[37]。此外,不同物種間每個細胞的RNA含量相差很大,哺乳動物細胞含有大約20~25 pg RNA,而真菌細胞和細菌分別為0.5~1 pg和0.05~0.1 pg[38],因此真菌和細菌RNA濃度高低通常是影響轉錄組分析的一個重要因素。此外讀數覆蓋率也是一個重要因素,Dual RNA-seq的有效讀取數一般在50~100 bp,一般讀取數越長,在基因組中的定位越準確。冗余讀數也會存在影響,主要表現在rRNA感染細胞時高達98%,是冗余讀數的重要來源[39],可以通過剔除rRNA或者在mRNA尾部添加poly(A)過濾掉rRNA,殘余的rRNA,讀取數可以使用SortMeRNA工具在計算機上進行刪除[40]。
相信隨著測序技術不斷發展,高通量測序的深度必定會進一步提高,Dual RNA-seq技術也會越來越成熟的應用于更多領域研究。比如對于新發傳染病的病原體而言,通過NGS技術測的序列往往比較新,與己知參考序列差異較大,通過傳統的檢測手段很難對其做出準確的判斷[5],而基于NGS技術的Dual RNA-seq不僅可以及時對其做出判斷,同時還能了解和追蹤病原體和宿主在感染過程中發生的生理及病理變化,為診治新發傳染病提供一定的依據;Dual RNA-seq也可不依賴病毒培養,對于那些臨床難以培養的病毒的研究顯得尤其關鍵;此外,RNA病毒的基因組由核糖核苷酸組成,Dual RNA-seq技術可以在核苷酸水平平行比較病毒感染后組織與細胞中差異表達的基因,從而更直觀,準確的揭示病毒的嗜性和改變病程,從而理解病毒與宿主之間的相互作用[41-42]。因此利用 Dual RNA-seq 技術開展病毒與宿主轉錄組間基因表達的研究,分析病毒感染后病毒與宿主轉錄組水平的基因調控應答特征,鑒定與致病性相關的關鍵基因,進一步在生物學角度闡明病毒的分子致病機制提供科學的證據,也可以作為一種診斷方法,對未知疾病進行診斷。本文針對Dual RNA-seq在各宿主-病原體相互作用進行綜述,相信Dual RNA-seq技術優勢將會在轉錄組學研究中有廣闊的應用基礎和前景,并為宿主-病原體相互作用機制的研究提供有力的技術支撐。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
體育科技文獻通報(2022年3期)2022-05-23 13:46:54
天津外國語大學學報(2021年3期)2021-08-13 08:32:18
遼金歷史與考古(2021年0期)2021-07-29 01:06:54
學苑創造·A版(2020年9期)2020-10-13 09:41:02
科技傳播(2019年22期)2020-01-14 03:06:54
民用飛機設計與研究(2019年4期)2019-05-21 07:21:24
小學生學習指導(低年級)(2017年10期)2017-10-10 01:00:05
汽車工程學報(2017年2期)2017-07-05 08:13:02
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
