氣相色譜法測定食用植物油中有機氯類農(nóng)藥殘留
2022-10-29 06:07:32任鵬柴艷兵張耀廣張洪鑫李興佳白曉云王鴿董新明
食品研究與開發(fā) 2022年20期
任鵬,柴艷兵,張耀廣,張洪鑫,李興佳,白曉云,王鴿*,董新明
(1.天津食品集團有限公司,天津 300000;2.君樂寶乳業(yè)集團農(nóng)業(yè)農(nóng)村部乳品質(zhì)量安全控制重點實驗室,河北 石家莊 050021;3.天津康復(fù)療養(yǎng)中心,天津 300191)
有機氯農(nóng)藥主要成分為苯和環(huán)戊二烯兩類[1],用于防治植物疾病與蟲害等,其作用明顯但具有高毒性、低揮發(fā)性、不易降解、高生物積累性等缺點[2],易溶于脂肪和有機溶劑,會通過食物鏈的放大作用,對人類健康造成危害[3],主要損害中樞神經(jīng)和肝、腎等。而造成以上損害的主要原因則是中毒,中毒類型分為急性中毒和慢性中毒兩種[4-6]。急性中毒癥狀有頭痛、眩暈、惡心、嘔吐、流涎、肌無力、肌震顫等,嚴重者有大汗淋漓、發(fā)熱、渾身震顫、抽搐、昏迷等癥狀[7-8]。慢性中毒癥狀常表現(xiàn)為神經(jīng)衰弱綜合征。慢性中毒的毒理作用主要表現(xiàn)在對神經(jīng)系統(tǒng)、內(nèi)分泌系統(tǒng)造成影響并侵害肝臟、腎臟等器官[9-11]。
李曼曼等[12]建立了一種利用QuEChERS(quick、easy、cheap、effective、rugged、safe) 結(jié)合氣相色譜-三重四極桿串聯(lián)質(zhì)譜檢測畜禽肉中17種有機氯農(nóng)藥殘留的方法。結(jié)果顯示17種有機氯農(nóng)藥在1 μg/kg~100 μg/kg范圍內(nèi)線性關(guān)系良好,檢測限為 0.07 μg/kg~0.31μg/kg。王曉春等[13]建立了蔬菜及水果中16種有機氯農(nóng)藥殘留的氣相色譜檢測方法,檢出限在0.16 μg/kg~2.90 μg/L范圍內(nèi),在4種基質(zhì)中的加標回收率為70.1%~119%,相對標準偏差為0.23%~5.2%。黃小波等[14]建立了QuEChERS方法測定雞蛋中17種農(nóng)藥的殘留,結(jié)果表明17種農(nóng)藥在0~0.100 μg/mL范圍內(nèi),線性關(guān)系良好,相關(guān)系數(shù)R2在0.996 4~0.999 9,加標回收率為76.3%~102%,方法檢出限在0.01μg/kg~0.28μg/kg。李妃等[15]建立了海水中16種有機氯農(nóng)藥的氣相色譜測定方法,樣品經(jīng)前處理后利用電子俘獲檢測器(electron capture detector,ECD)進行分析測定,色譜系統(tǒng)采用DB-1701毛細管色譜柱,結(jié)果顯示在0.75 μg/L~200 μg/L范圍內(nèi),16種有機氯農(nóng)藥具有良好的線性關(guān)系,檢出限在0.53 ng/L~2.90 ng/L之間,加標回收率為82.0%~107.9%。李福敏等[16]建立了氣相色譜-電子捕獲檢測器方法檢測大米中32種有機氯類農(nóng)藥殘留量的分析方法,結(jié)果顯示32種目標物在優(yōu)化試驗條件下有較好的分離效果,方法檢出限為 1.0 μg/kg~20.0 μg/kg,定量限為 2.0 μg/kg~25.0 μg/kg。試驗相對標準偏差為 0.19%~8.6%(n=6);張靜靜[17]通過對吉林省土壤中有機氯類農(nóng)藥的分析,總結(jié)出長春市城郊菜地土壤中有機氯農(nóng)藥殘留主要為六六六和滴滴涕,兩者共占有機氯農(nóng)藥殘留總量的88.29%;吉林市城郊菜地土壤中有機氯農(nóng)藥殘留主要為六六六和滴滴涕,兩者共占有機氯農(nóng)藥殘留總量的82.05%;檢測城市蔬菜依然有有機氯類農(nóng)藥殘留存在。
農(nóng)藥中有機殺蟲劑包括有機磷類、有機氯類、氨基甲酸酯類、擬除蟲菊酯類、特異性殺蟲劑等。我國于1983年已經(jīng)禁止有機氯類農(nóng)藥的使用與生產(chǎn)[18],但該類農(nóng)藥在土壤與植物中的檢出率依然較高。植物是食用植物油的主要原料[19-20],因此食用植物油中存在有機氯農(nóng)藥殘留的風險,然而食用植物油和一般食品基質(zhì)差異較大,主要由脂肪酸甘油酯組成,而大多數(shù)農(nóng)藥是脂溶性的,所以檢測食用植物油中的農(nóng)藥殘留時,必須除去基質(zhì)中的脂肪、色素及其他相對分子質(zhì)量較高的干擾物,以免檢測儀器受到基質(zhì)干擾物的污染,導(dǎo)致儀器使用壽命變短。對于食用植物油中殘留農(nóng)藥的檢測,常用的液-液萃取和固相萃取等前處理步驟繁瑣,試劑使用量大,前處理時間長,檢測大批量樣品時效率低。而QuEChERS法以其快速、簡便、價格低廉、有效、可靠和安全的優(yōu)點在農(nóng)藥殘留分析中得到了廣泛應(yīng)用,并已用于蔬菜、大米、水果、茶葉等多種食品基質(zhì)中。
目前,任雅君等[21]整體介紹了現(xiàn)階段國內(nèi)主要的植物油中農(nóng)藥殘留的前處理方法以及常用的檢測方法(氣質(zhì)聯(lián)用和液質(zhì)聯(lián)用);GB/T 5009.19—2008《食品中有機氯農(nóng)藥多組分殘留量的測定》和GB/T 5009.162—2008《動物性食品中有機氯農(nóng)藥和擬除蟲菊酯農(nóng)藥多組分殘留量的測定》中提出檢測食用油采用氣相色譜法[22-23],但以上方法的前處理操作均用到大量的石油醚、乙酸乙酯等毒性較大的有機試劑,因此,本研究綜合借鑒QuEChERS快速前處理方法,以大豆油為代表性基質(zhì),采用N-丙基乙二胺(primary secondary amine,PSA)和C18粉末作為QuEChERS法的吸附劑去除食用植物油基質(zhì)中的干擾物,結(jié)合氣相色譜建立快速、高效的食用植物油中18種有機氯農(nóng)藥的檢測方法,為今后食用植物油中檢測有機氯農(nóng)藥,提供理論依據(jù)和技術(shù)支撐。
1 材料與方法
1.1 材料與試劑
大豆油(5 L):市售。
18 種農(nóng)藥殘留標準品[α-六六六、β-六六六、γ-六六六、δ-六六六、七氯、艾氏劑、氧氯丹、環(huán)氧七氯、反-氯丹、α-硫丹、順-氯丹、狄氏劑、p,p'-DDE,2,2-雙(對氯苯基)-1-氯乙烯、β-硫丹、p,p'-DDD、o,p'-DDT、硫丹硫酸鹽、p,p'-DDT]:農(nóng)業(yè)部環(huán)境保護科研研究所;無水硫酸鎂:天津市永大化學試劑有限公司;乙腈(色譜純):默克化工技術(shù)(上海)有限公司;N-丙基乙二胺填料(PSA填料)、C18填料:天津一方科技有限公司;正己烷(色譜純):北京迪科馬科技有限公司。
1.2 儀器與設(shè)備
色譜柱(HP-5,柱長 30 m,內(nèi)徑 0.32 mm,膜厚0.25 μm):安捷倫科技(中國)有限公司;氣相色譜儀(8890,配電子俘獲檢測器):島津儀器蘇州有限公司;分析天平(BT25S):德國賽多利斯股份公司;渦旋混合器(IKA-MS3):德國IKA有限公司;旋轉(zhuǎn)蒸發(fā)儀(BM510C):日本 Yamato公司;離心機(TGL-20M):上海盧湘儀離心機儀器有限公司。
1.3 試驗方法
1.3.1 標準工作液的配制
準確移取18種農(nóng)藥殘留標準品(100 μg/mL)1 mL,用正己烷溶解并定容至10 mL,此為標準儲備液(10 μg/mL);準確移取標準儲備液 1 mL,用正己烷溶解并定容至 10 mL,此為標準中間液(1 μg/mL);準確移取標準中間液1 mL,用正己烷溶解并定容至10 mL,此為標準使用液(100 ng/mL)。
本方法確認所做曲線的工作范圍為(5 ng/mL~60 ng/mL),分別吸取標準使用液 50、100、200、300、500、600 μL 用正己烷定容至 1 mL,渦旋混勻,配制成濃度為 5.00、10.00、20.00、30.00、50.00、60.00 ng/mL 的標準工作液。以峰面積為縱坐標,以農(nóng)藥殘留各組分含量為橫坐標,繪制標準曲線。
1.3.2 儀器條件
檢測器類型為ECD,載氣:高純氮氣,恒壓模式,進樣口溫度280℃,檢測器溫度300℃,進樣體積1 μL,不分流,電流保持 1.0 nA,程序升溫:60 ℃保持1 min,以40℃/min升至170℃,以1.2℃/min升至230℃,再以40℃/min升至280℃,保持5 min。
1.3.3 樣品處理
稱取2 g樣品于50 mL離心管中,加入15 mL乙腈,渦旋混勻,8 000 r/min離心5 min。用吸管將有機相乙腈全部轉(zhuǎn)移到另一離心管中后,重復(fù)提取一次,合并提取液。向30 mL提取液中加入10 mL用乙腈飽和的正己烷,除去乙腈中的油脂,渦旋混勻,5 000 r/min離心5 min,取下層乙腈于新的離心管中,加入0.2 g PSA填料,0.2 g C18填料、2 g無水硫酸鎂,渦旋混勻,8 000 r/min離心5 min,轉(zhuǎn)移上清液至旋蒸瓶中,旋轉(zhuǎn)蒸干,加入1.00 mL正己烷,在渦旋混合器上充分振蕩溶解殘留物,經(jīng)0.22 μm有機系濾膜過濾后,采用氣相色譜檢測。
1.3.4 色譜條件優(yōu)化
在保持唯一變量的情況下,固定壓力模式、檢測器溫度、進樣體積,電流保持1.0 nA,程序升溫:60℃保持1 min,以40℃/min升至170℃,以1.2℃/min升至230℃,再以40℃/min升至280℃,保持5 min,更改分流比、進樣口溫度進行上機測試。
1.4 數(shù)據(jù)處理
每組數(shù)據(jù)均重復(fù)3次,采用WPS Office11.1.0.11372-release軟件進行數(shù)據(jù)統(tǒng)計分析,使用Origin2017軟件繪制結(jié)果圖。
2 結(jié)果與分析
2.1 色譜條件的優(yōu)化及標準曲線
進樣口溫度是樣品氣化溫度,影響化合物氣化的關(guān)鍵因素,在保持唯一變量的情況下,不同進樣口溫度對儀器響應(yīng)值的影響如圖1所示;分流比直接影響進入儀器的氣體量及響應(yīng)值結(jié)果,不同分流比對響應(yīng)值的影響如圖2所示。

圖1 不同進樣口溫度對響應(yīng)值的影響Fig.1 Influence of inlet temperature on response
由圖1可知,進樣口溫度為280℃的18種有機氯農(nóng)藥均有最大響應(yīng)值,由圖2可知,除p,p'-DDT外其余17種有機氯農(nóng)藥在不分流情況下響應(yīng)值最大。最終確定儀器條件:載氣為高純氮氣、恒壓模式、進樣口溫度280℃、檢測器溫度ECD 300℃、進樣體積1 μL、不分流進樣、電流1.0 nA。程序升溫至60℃保持1 min,以40℃/min升至170℃,以1.2℃/min升至230℃,再以40℃/min升至280℃,保持5 min。
18種有機氯農(nóng)藥分離色譜圖如圖3所示。
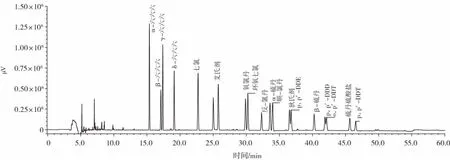
圖3 18種有機氯農(nóng)藥色譜圖Fig.3 Chromatography of 18 organochlorine pesticides
由圖3可知,18種有機氯農(nóng)藥出峰峰形對稱,無雜質(zhì)峰干擾,分離效果較好。所選取濃度為5.00、10.00、20.00、30.00、50.00、60.00 ng/mL,以峰面積為縱坐標,以農(nóng)藥殘留各組分含量為橫坐標,繪制標準曲線,試驗表明,農(nóng)藥殘留各組分的含量在0~60 ng/mL內(nèi)呈線性,具體數(shù)值見表1。

表1 農(nóng)藥殘留各組分線性分析Table 1 Linearity of each pesticide residue

續(xù)表1 農(nóng)藥殘留各組分線性情況Continue table 1 Linearity of each pesticide residue
從表1可以看出,18種農(nóng)藥殘留混標的各組分線性均大于0.990,符合GB/T27404—2008《實驗室質(zhì)量控制規(guī)范食品理化檢測》中對校準曲線相關(guān)系數(shù)的要求。
2.2 前處理方法優(yōu)化
2.2.1 提取溶劑的確定
分別采用甲醇、乙腈、50%的甲醇乙腈溶液做提取試劑,保持相同提取溶劑體積,用甲醇和甲醇乙腈混合溶液提取時,回收率在45%以下,不滿足標準要求,使用乙腈提取樣品時回收率達到90%,所以試驗選取乙腈作為提取試劑。
2.2.2 除脂試劑的確定
不同除脂試劑的加標回收率見表2。

表2 不同除脂試劑的加標回收率Table 2 Spiked recoveries of different degreasing reagents
乙腈提取大豆油后,部分脂類物質(zhì)溶于乙腈,造成提取液不能完全蒸發(fā)至干,影響定容體積。因正己烷具有除脂效果,故在提取液中加入10 mL正己烷,結(jié)果顯示使用純正己烷,除脂效果明顯,但試驗回收率低,分析原因是正己烷能溶解部分乙腈,嘗試加入用乙腈飽和的正己烷,回收率達到標準要求,所以試驗選用乙腈飽和的正己烷作為除脂試劑。
2.2.3 正確度與精密度
選擇3個不同添加水平進行加標回收率試驗,3 個水平的加標量分別為 0.005、0.010、0.020 mg/kg,分別做6個平行,結(jié)果見表3。
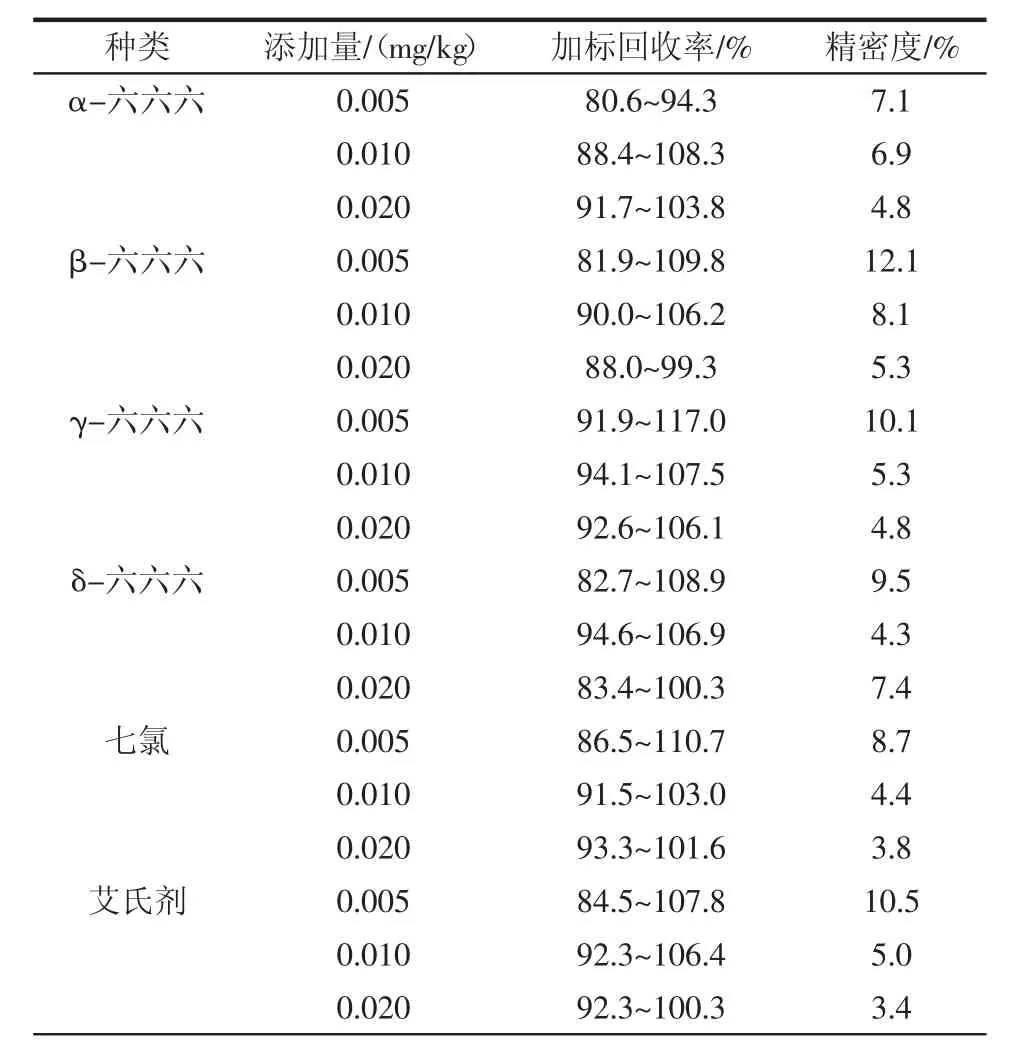
表3 加標回收率Table 3 Spiked recovery
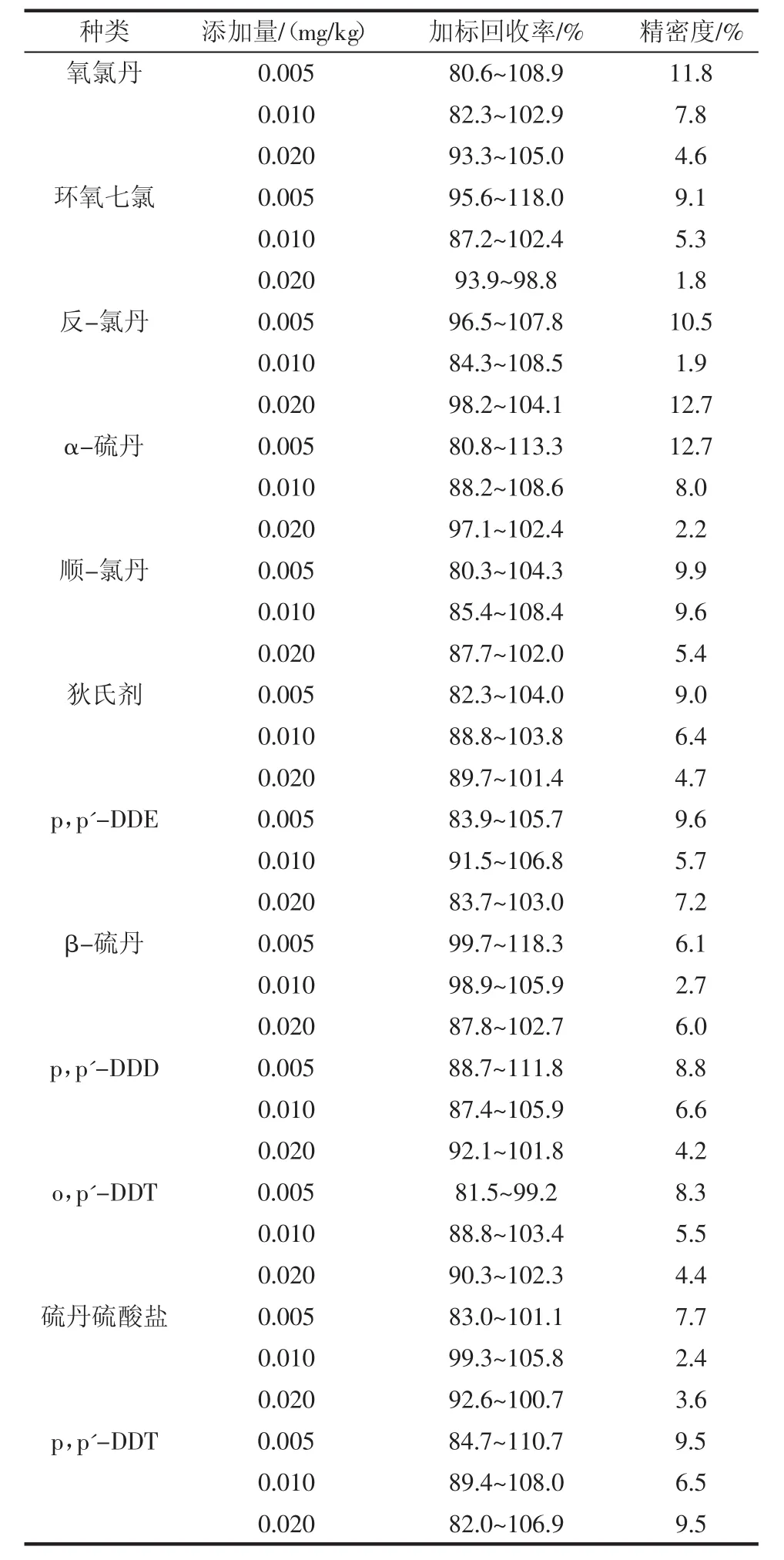
續(xù)表3 加標回收率Continue table 3 Spiked recovery
通過表3可以看出,在大豆油基質(zhì)中,18種有機氯農(nóng)藥加標回收率為80.3%~118.3%。各組分加標回收率可以滿足實驗室對于大豆油中有機氯農(nóng)藥殘留篩查的要求,所得結(jié)果的變異系數(shù)為1.8%~12.7%,滿足GB/T 27404—2008《實驗室質(zhì)量控制規(guī)范食品理化檢測》中變異系數(shù)要求。結(jié)果顯示該方法有較好的準確度和精密度,可以作為檢測大豆油中有機氯農(nóng)藥殘留的方法。
3 結(jié)論
本文通過優(yōu)化進樣口溫度、分流比,選取乙腈作為提取試劑、乙腈飽和的正己烷作為除脂試劑建立了一種氣相色譜法檢測食用植物油中有機氯農(nóng)藥殘留的方法,數(shù)據(jù)表明該方法提取、除脂效果好,分離度高,適用于脂肪含量高的油類物質(zhì)。該法操作簡單,準確度、靈敏度高,可以滿足同時檢測食用植物油中18種有機氯農(nóng)藥的要求,為檢測食用植物油中其他種類農(nóng)藥殘留提供理論依據(jù)。
猜你喜歡
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2021年6期)2021-11-22 07:50:58
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
中學生數(shù)理化·七年級數(shù)學人教版(2020年12期)2021-01-18 06:57:46
兒童故事畫報(2019年5期)2019-05-26 14:26:14
海峽科技與產(chǎn)業(yè)(2016年3期)2016-05-17 04:32:12
Coco薇(2016年2期)2016-03-22 02:42:52
Coco薇(2015年1期)2015-08-13 02:47:34
小雪花·成長指南(2015年7期)2015-08-11 15:03:12
