氣相色譜-串聯質譜法測定植物源化妝品中53種農藥殘留
2022-11-18 10:07:04梁梓洋梁梓豪馬葉芬羅輝泰吳惠勤
分析測試學報 2022年11期
梁梓洋,梁梓豪,馬葉芬,羅輝泰,黃 芳,吳惠勤
(廣東省科學院測試分析研究所(中國廣州分析測試中心),廣東省化學測量與應急檢測技術重點實驗室,廣東省中藥質量安全工程技術研究中心,廣東 廣州 510070)
近年來,隨著社會經濟的發展,人們的生活水平穩步提高,對化妝品的要求也隨之不斷提高。《中華人民共和國國民經濟和社會發展第十四個五年規劃和2035年遠景目標綱要》明確指出,要開展中國品牌創建行動,保護發展中華老字號,提升自主品牌影響力和競爭力,率先在化妝品、服裝、家紡、電子產品等消費品領域培育一批高端品牌[1],這標志著我國化妝品行業科技引領的高質量發展時代已到來。在市場潮流的推動下,綠色化及具有特定功效的化妝品日益受到青睞,許多化妝品以添加天然植物成分為亮點,植物提取物在化妝品中的應用日趨廣泛[2]。植物提取物的引入不僅可為化妝品帶來獨特香味,其活性成分還具有滋養、修護、祛痘等特定功效[3],因此植物提取物的正確應用能為化妝品帶來更好的美容護膚效果[4],具有廣闊的應用前景。但由于環境污染或種植過程中非法使用農藥等問題[5-6],在化妝品引入植物提取物原料時會帶來農藥殘留的安全風險。
由農藥殘留引起的化妝品安全問題已逐漸得到國內外研究者的高度重視。早在2010年,國家食品藥品監督管理總局在《關于印發化妝品中可能存在的安全性風險物質風險評估指南的通知》中指出,使用植物來源原料的化妝品應說明其農藥殘留的情況。2013年,歐盟在化妝品法規EC1223/2009中明確指出艾氏劑、狄氏劑等農藥屬于化妝品中的禁用組分[7];我國《化妝品安全技術規范(2015年版)》中明確規定多種農藥成分為化妝品的禁用組分[8]。2020年,國家市場監督管理總局發布了《含植物提取物類化妝品中55種禁用農藥殘留量的測定》(GB/T 39665-2020),為植物源化妝品的農藥殘留檢測提供了依據[9]。
目前,化妝品中農藥殘留的測定方法主要有液相色譜法、氣相色譜法、液相色譜-串聯質譜法和氣相色譜-質譜法等[7,10-12]。GB/T 39665-2020中使用氣相色譜-質譜法及液相色譜-串聯質譜法進行測定,前者可測定35種禁用農藥。然而植物源化妝品基質比傳統化妝品更為復雜,使用氣相色譜-質譜法的選擇離子掃描模式(SIM)也難以避免干擾。而串聯質譜(MS/MS)可對母離子進行選擇性分離,利用其產生的子離子進行檢測,可提高分辨能力并大大降低背景值,采用氣相色譜-串聯質譜法的選擇反應監測模式(SRM)可在復雜基質情況下獲得更好的選擇性和抗干擾能力[13-15],從而提高分析的靈敏度,近年來被越來越多地用于多種基質的禁用農藥殘留檢測中[16-19]。例如《中國藥典(2020年版)》[20]中,規定使用氣相色譜-串聯質譜法及液相色譜-串聯質譜法對中藥材中33種禁用農藥進行測定。植物源化妝品的基質復雜程度更高,但尚未見使用氣相色譜-串聯質譜法測定其農藥殘留的報道。本文選取GB/T 39665-2020和《中國藥典(2020年版)》中涉及的53種禁用農藥,以水基型的精華液和非水基型的膏霜2種代表性樣品為研究對象優化樣品前處理方法,建立了植物源化妝品中53種禁用農藥殘留的氣相色譜-串聯質譜測定方法。該方法樣品前處理簡單快速,方法靈敏度和選擇性高,重復性好,適用于化妝品中多種農藥殘留的同時、快速檢測,為保障化妝品質量安全和促進我國化妝品的高質量發展提供了技術支撐。
1 實驗部分
1.1 儀器與設備
氣相色譜-三重四極桿質譜聯用儀(TRACE 1300-TSQ 9000,美國Thermo Fisher Scientific公司);超聲波清洗器(東莞市超聲波設備有限公司);電子天平(美國Sartorious公司);渦旋振蕩器(德國IKA公司);QB-600型高速振蕩混合器(海門市其林貝爾儀器制造有限公司);氮吹濃縮儀(青島聚創環保集團有限公司);冷凍高速離心機(湘儀集團);Captiva EMR-Lipid固相萃取柱(300 mg/3 mL,美國Agilent公司);0.22 μm有機相濾膜(美國Agilent公司)。
1.2 材料與試劑
53種農藥對照品(壇墨質檢標準物質中心);乙腈、丙酮、乙酸乙酯(色譜純,德國Merck公司);實驗用水為二次蒸餾水;氯化鈉(分析純,廣州化學試劑廠)。
1.3 標準溶液的配制
1.3.1 標準儲備溶液與工作溶液的配制分別準確稱取適量待測農藥對照品,用乙腈配制成質量濃度為1 mg/mL的標準儲備溶液。使用時,根據需求用乙腈稀釋成不同質量濃度的標準工作溶液。
1.3.2 空白樣品基質溶液與基質混合標準工作溶液的配制稱取經測定不含目標待測物的空白樣品0.5 g,按照樣品前處理方法制得空白樣品基質溶液。經氮氣濃縮后,加入不同質量濃度的標準工作溶液后定容,得到基質混合標準工作溶液。
1.4 樣品前處理
液體樣品(精華液、化妝水及原液等):稱取試樣0.5 g(精確至0.000 1 g)于15 mL離心管中,加入3 mL飽和氯化鈉溶液稀釋并混勻,隨后加入5 mL乙腈,渦旋混合30 s后以1 120次/min的頻率振蕩提取10 min,以10 000 r/min離心3 min。取上清液于另一15 mL離心管中,剩余部分用5 mL乙腈再提取1次,合并兩次提取液,于35℃水浴下氮吹濃縮至約3 mL。濃縮后的提取液過EMR固相萃取柱凈化。收集洗脫液于35℃水浴下氮吹至約0.3 mL,用乙腈定容至0.5 mL,經有機相濾膜過濾后待測定。
膏霜和乳液樣品(面霜、精華霜及護發乳等):稱取試樣0.5 g(精確至0.000 1 g)于15 mL離心管中,加入3 mL飽和氯化鈉溶液后渦旋混合1 min,超聲處理5 min使樣品充分分散。加入5 mL乙腈于上述離心管中,渦旋混合30 s后以1 120次/min的頻率振蕩提取10 min,以10 000 r/min離心3 min。取上清液于另一15 mL離心管中,剩余部分用5 mL乙腈再提取1次,合并兩次提取液,于35℃水浴下氮吹濃縮至約3 mL。置于-24℃下冷凍20 min,于4℃下以10 000 r/min離心2 min,上清液過EMR固相萃取柱凈化。收集洗脫液于35℃水浴下氮吹至約0.3 mL,用乙腈定容至0.5 mL,經有機相濾膜過濾后待測定。
1.5 實驗條件
1.5.1 色譜條件色譜柱:Agilent VF-17ms毛細管柱(30 m×0.25 mm×0.25 μm);色譜柱升溫程序:70℃保持2 min,以25℃/min升至150℃,以3℃/min升至200℃,再以8℃/min升至280℃保持10 min;載氣:氦氣(純度≥99.999%);載氣流速:1.0 mL/min;進樣口溫度:260℃;進樣量:1 μL;進樣方式:脈沖不分流進樣,脈沖壓力為150 kPa,0.75 min后打開分流閥和隔墊吹掃。
1.5.2 質譜條件電子轟擊電離源(70 eV);離子源溫度:280℃;傳輸線溫度:280℃;碰撞氣:氬氣;掃描方式:選擇反應監測(SRM)模式,時間窗口為保留時間±1.0 min。53種農藥的定性定量離子對、碰撞能量等質譜參數見表1。
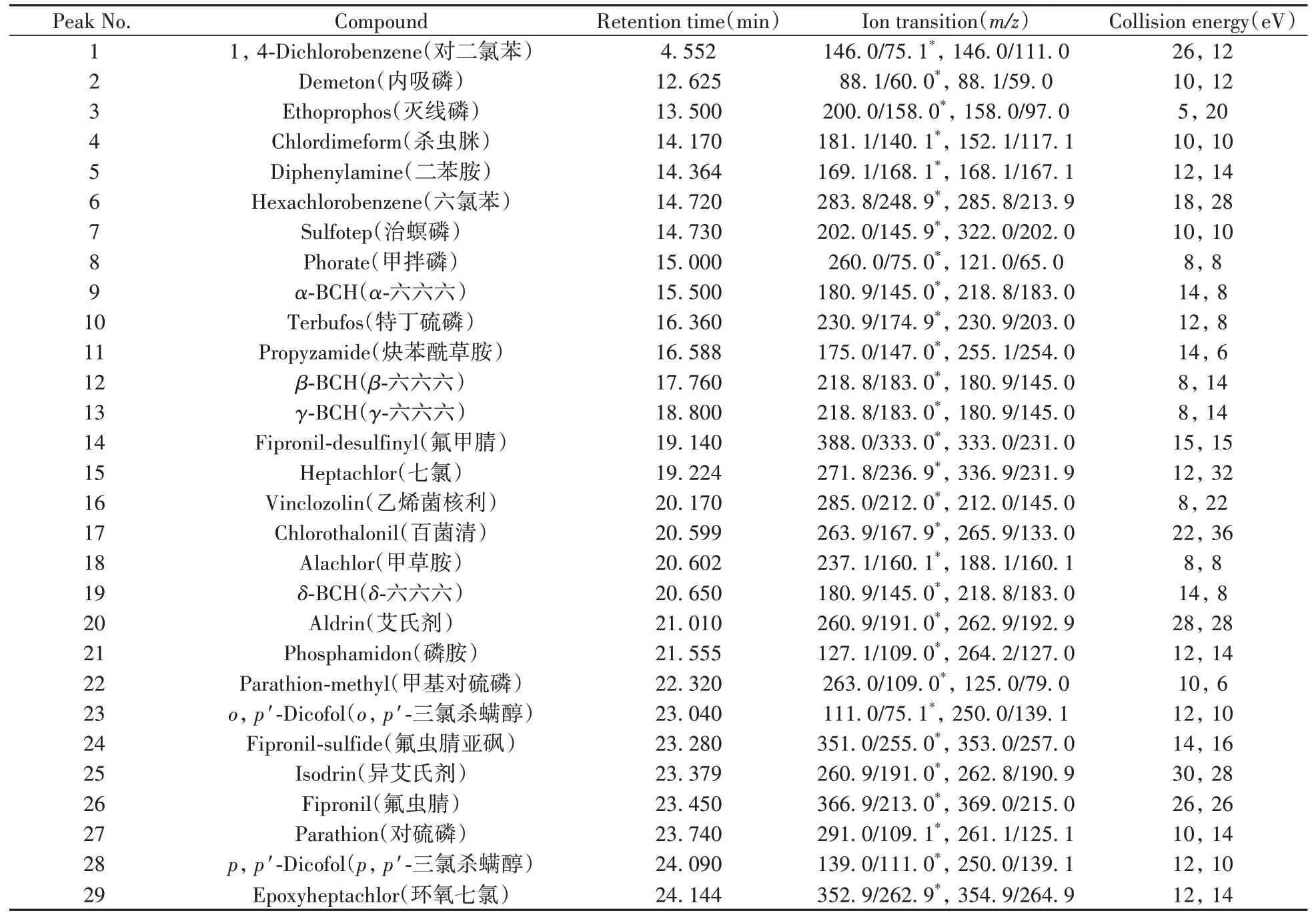
表1 53種農藥的質譜參數Table 1 Mass spectrometry parameters of 53 pesticides
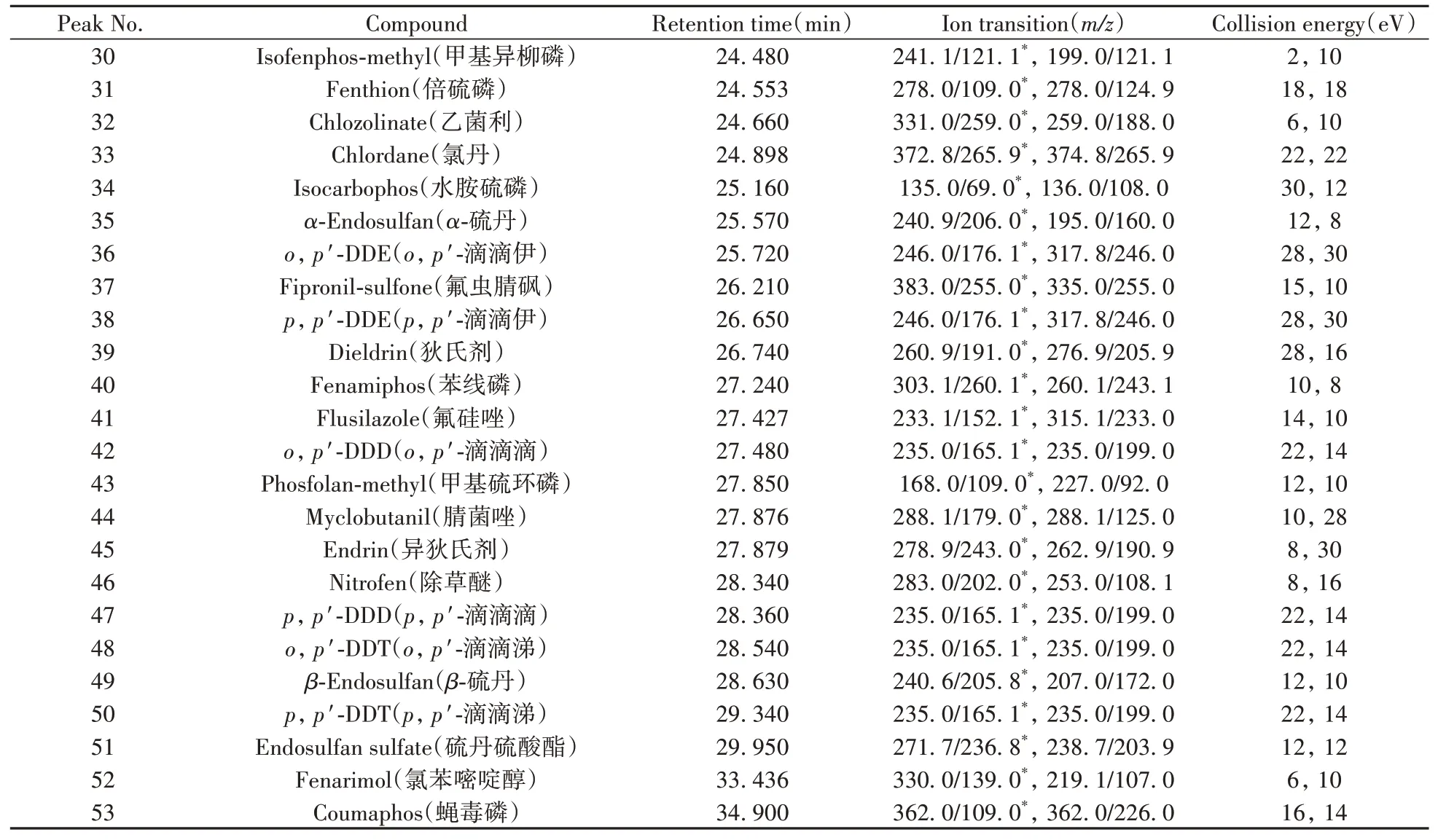
(續表1)
2 結果與討論
2.1 儀器分析條件的優化
2.1.1 色譜柱的選擇分別考察了弱極性的DB-5MS(30 m×0.25 mm×0.25 μm)和中等極性的VF-17ms(30 m×0.25 mm×0.25 μm)兩種色譜柱的分離效果。實驗表明,兩種色譜柱均能使53種農藥實現基線分離,對于互為異構體的農藥,如β-六六六/γ-六六六和p,p'-滴滴滴/o,p'-滴滴滴,在VF-17ms色譜柱上能夠取得更滿意的分離度。因此,本實驗選擇VF-17ms柱作為色譜柱。
2.1.2 質譜條件的優化使用目標物的混合標準工作溶液,對53種農藥進行質譜全掃描分析,在各化合物的一級質譜中選取豐度較高的分子離子峰或質荷比較大的基峰作為母離子,對各化合物的母離子施加一定的碰撞能量獲得其二級質譜,選取豐度較高且質荷比較大的碎片離子作為子離子。通過優化碰撞能量使53種農藥的離子對強度達到最大值,從而確定各化合物最優的質譜參數,如“1.5.2”所示。53種農藥在優化條件下的SRM總離子流圖見圖1。
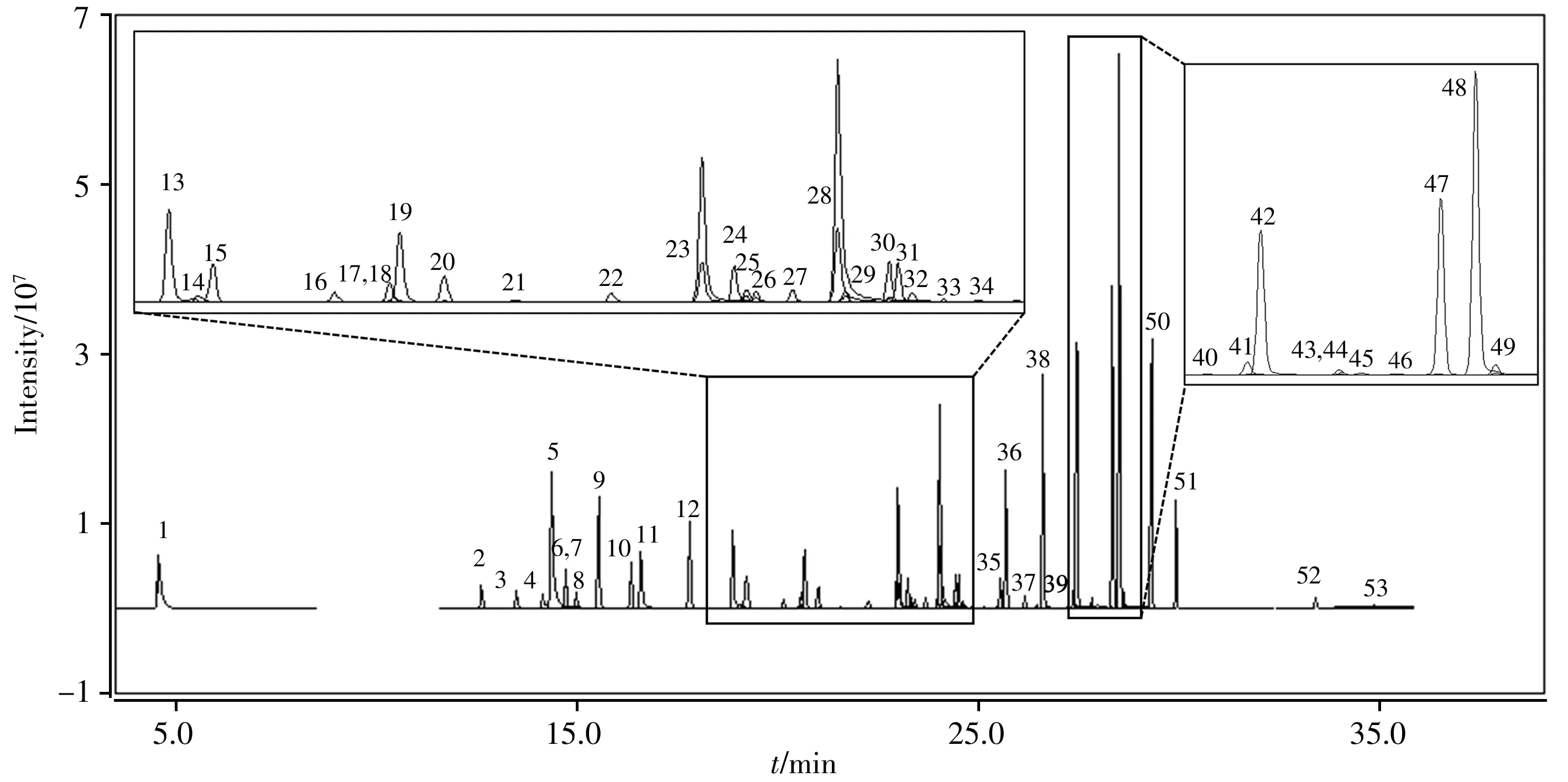
圖1 53種農藥在選擇反應監測模式掃描下的總離子流圖Fig.1 Total ion chromatograms of 53 pesticides using SRM mode peak numbers 1 to 53 are corresponding with the compounds shown in Table 1
2.2 前處理條件的優化
2.2.1 提取溶劑的選擇由于化妝品基質較復雜,不同種類的農藥性質也存在差異,提取效率易受到基質的影響,因此需選擇提取效率高且對基質提取較少的溶劑作為提取溶劑。常見的提取溶劑主要有乙腈、丙酮和乙酸乙酯等[21]。本文以精華液和膏霜樣品為研究對象,考察了乙腈、丙酮和乙酸乙酯對目標物的提取效率(見表2)。對于部分膏霜樣品,需先加入飽和氯化鈉溶液進行破乳和分散,提高提取溶劑的提取效率;對于精華液樣品,加入飽和食鹽水可降低樣品的黏度,同時可使有機相與水相更好地分層。結果表明,3種提取溶劑均能從精華液和膏霜兩種基質中提取53種目標農藥,但由于丙酮與乙酸乙酯的極性相對較小,在提取目標化合物的同時也提取了膏霜樣品中的大量油脂,會對進樣口和色譜柱造成污染[22]。而乙腈的極性適中,且能夠與飽和氯化鈉溶液較好地分層,對53種目標化合物的提取效率也較高,因此選擇乙腈作為提取溶劑。
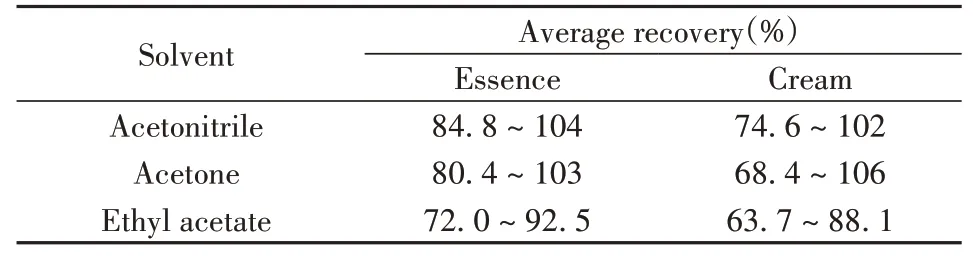
表2 不同溶劑對53種農藥的提取效率Table 2 Extraction efficiencies of 53 pesticides with different solvents
2.2.2 提取方式的選擇比較了渦旋提取和振蕩提取兩種方式的提取效果。結果表明,以乙腈作為提取溶劑,采用渦旋提取時,53種農藥在精華液和膏霜樣品中的平均回收率分別為82.3%~104%和62.8%~102%;當采用振蕩提取時,53種農藥在精華液和膏霜樣品中的平均回收率分別為83.1%~102%和79.0%~108%,可見振蕩提取的效果較好。由于精華液和膏霜樣品具有一定的黏度,即使加入飽和氯化鈉溶液進行稀釋,其黏度仍較大,導致提取溶劑與樣品難以充分接觸。相比之下,利用振蕩提取可使兩相充分接觸,獲得相對較好的提取效率。綜合考慮,選擇乙腈振蕩提取作為提取方式。
2.3 樣品凈化方式的選擇
由于膏霜樣品中含有大量油脂,即使選擇極性較大的乙腈作為提取溶劑,也不可避免地共提取出一部分油脂,而過多的油脂會使固相萃取柱的填料飽和,影響提取液的凈化效果,并對進樣口和色譜柱造成污染。由于油脂在低溫條件下易從乙腈中析出[23],因此選擇在-24℃下冷凍處理20 min,以去除乙腈提取液中的油脂。為考察冷凍除脂對53種農藥回收率的影響,比較了冷凍處理前后的平均回收率。結果表明,膏霜樣品經冷凍處理前、后的回收率分別為58.5%~118%和70.9%~113%,說明冷凍處理能夠去除部分油脂而起到凈化作用。同時比較了精華液樣品在冷凍處理前后回收率的變化,結果發現處理前、后的回收率相差甚微,因此對于精華液樣品可省略該步驟。
盡管冷凍除脂可起到一定的凈化效果,但乙腈提取液中仍含有一定量的油脂,而EMR-Lipid固相萃取柱可以選擇性地從高油脂含量樣品中保留脂質[24-25],將油脂從提取液中分離,從而達到凈化效果。為進一步消除油脂對后續測定的影響,選擇該固相萃取柱進行凈化。膏霜樣品的乙腈提取液經冷凍除脂后,以EMR-Lipid柱進行凈化前、后53種農藥的平均回收率分別為68.7%~114%和76.6%~104%,說明EMR-Lipid柱能夠減弱基質的影響。此外,考察了EMR-Lipid固相萃取柱對精華液樣品乙腈提取液的凈化效果,處理前、后53種農藥的平均回收率分別為76.0%~94.3%和82.8%~101%,表明EMR-Lipid柱起到了一定的凈化效果。綜上,選擇冷凍除脂結合EMR-Lipid固相萃取柱作為樣品凈化方式。
2.4 基質效應
質譜分析中存在的基質效應(ME)會對測定結果造成較大影響。分別以精華液和膏霜基質為對象,考察53種農藥在這兩類樣品中的基質效應。選取經測定不含目標待測物的空白樣品,將經提取和凈化后配制的基質匹配標準溶液與使用乙腈配制的相同質量濃度的標準溶液同時按照“1.5”條件進行分析,得到標準曲線,并按公式計算基質效應:ME(%)=100%×(A-B)/B[7]。式中,A和B分別為基質匹配標準曲線和溶劑標準曲線的斜率。當ME>0時,為基質增強效應;當ME<0時,為基質抑制效應。
53種農藥的響應值在基質溶液中均有不同程度的增大,在精華液樣品中的平均ME值為21.4%~130%,在膏霜樣品中的平均ME值為25.5%~148%,均表現為基質增強效應。為減弱基質效應對目標化合物的影響,樣品采用基質匹配標準曲線進行定量。
2.5 線性范圍、檢出限與定量下限
采用基質匹配的外標標準曲線法進行定量分析,按照“1.5”條件對系列基質匹配標準工作溶液進行測定,以各化合物的定量離子對峰面積(y)對各自的質量濃度(x)進行回歸分析,得到各組分的回歸方程與相關系數。并在空白樣品中添加一定濃度的53種農藥混合標準溶液,測定結果以3倍信噪比為檢出限(LOD),10倍信噪比為定量下限(LOQ)。結果表明,53種農藥在兩種化妝品基質中具有良好的線性關系,相關系數(r2)為0.996 1~0.999 9,LOD為0.01~0.02 mg/kg,LOQ為0.02~0.05 mg/kg(見表3)。
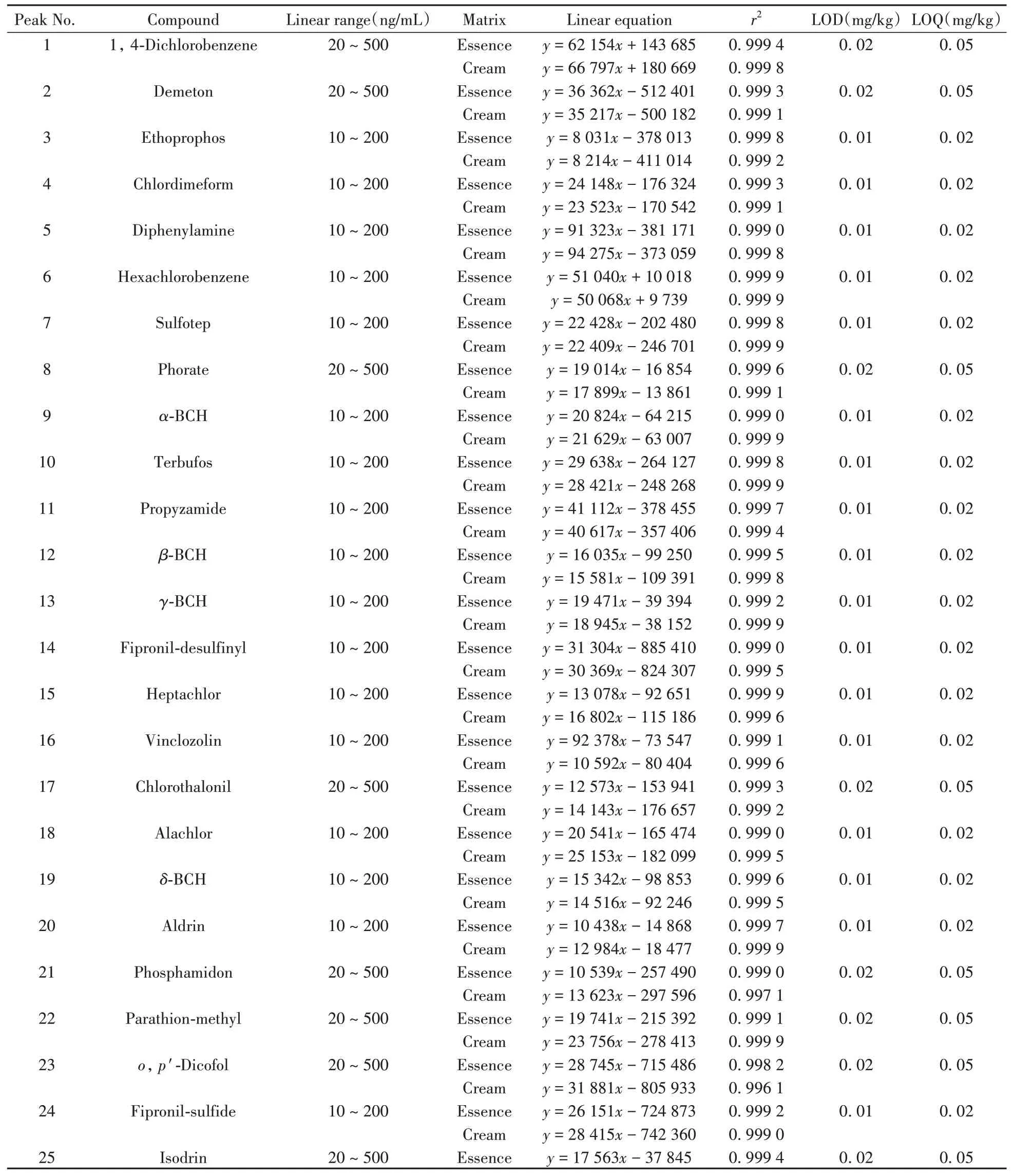
表3 53種農藥的線性范圍、線性方程、相關系數、檢出限和定量下限Table 3 Linear ranges,linear equations,correlation coefficients,LODs and LOQs for 53 pesticides
2.6 回收率與相對標準偏差
以精華液(水基樣品)和膏霜(非水基樣品)兩種化妝品作為代表性基質,選取經測定不含目標待測物的空白樣品進行加標回收率和精密度實驗。在0.05 mg/kg加標水平下進行6次重復實驗,以相應的基質匹配標準曲線計算53種農藥的回收率和相對標準偏差(RSD)。由表4可知,53種農藥在精華液樣品中的平均回收率為84.5%~102%,RSD為1.1%~4.9%;在膏霜樣品中的平均回收率為77.1%~105%,RSD為0.50%~4.0%,能夠達到GB/T 39665-2020的要求,滿足實際檢測需要。
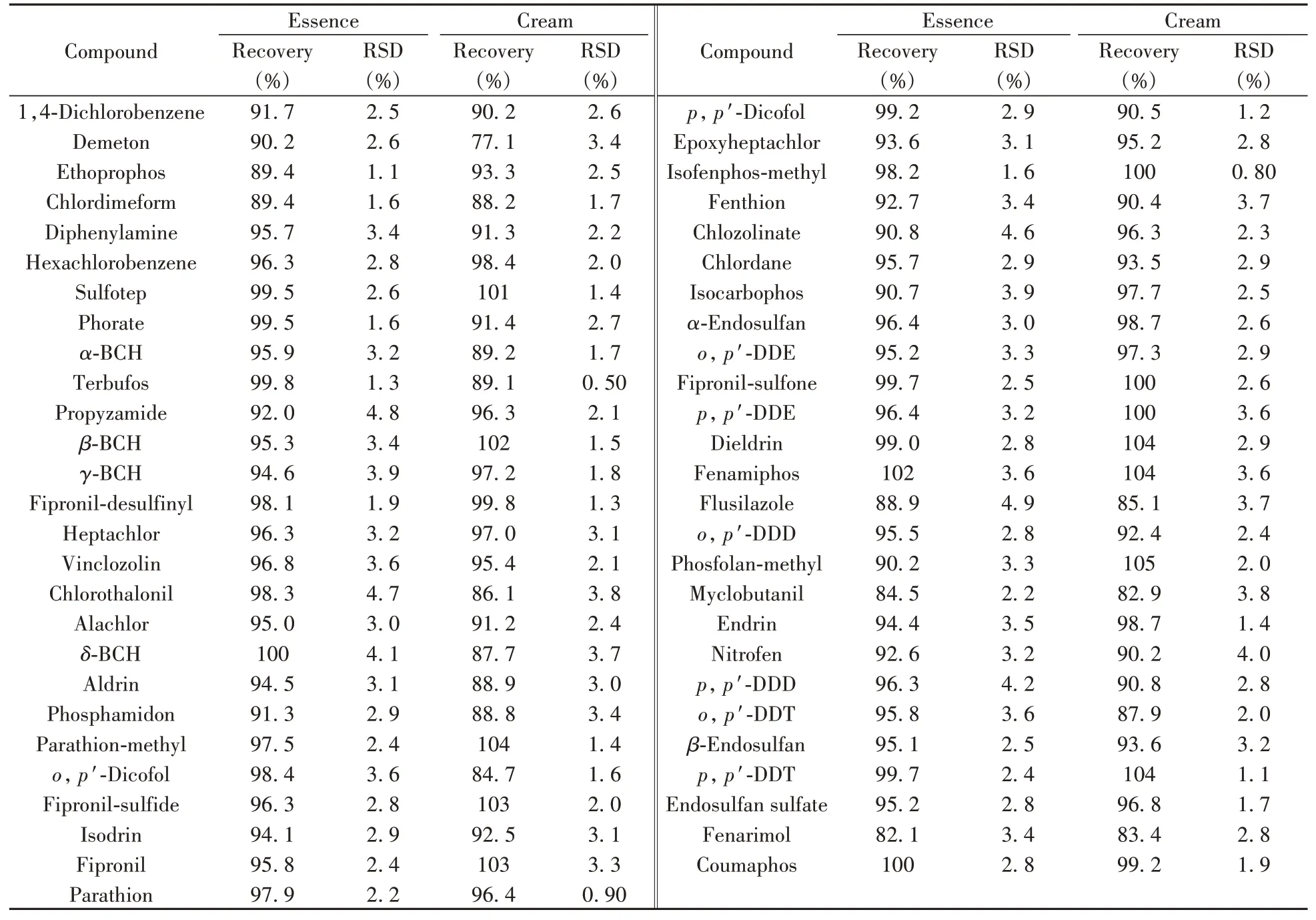
表4 精華液與膏霜樣品中53種農藥的加標回收率及相對標準偏差Table 4 Recoveries and relative standard deviations of 53 pesticides in essence and cream sample
2.7 實際樣品檢測
使用本方法對30個植物源化妝品進行檢測,檢出2個陽性樣品,在1個含人參提取物的膏霜樣品和另1個含金銀花提取物的液體樣品中,分別檢出0.06 mg/kg的六氯苯和0.04 mg/kg的氟蟲腈,陽性樣品的總離子流圖如圖2所示。盡管在化妝品中由植物提取物所引入的農藥殘留量一般較低,但由于大部分化妝品屬于日常使用的生活必需品,而在長期低劑量暴露于包括六氯苯在內的有機氯農藥及有機磷農藥情況下,會帶來生殖毒性及神經毒性等潛在安全風險[26-28],因此化妝品生產企業應重視植物提取物原料中農藥殘留的控制。
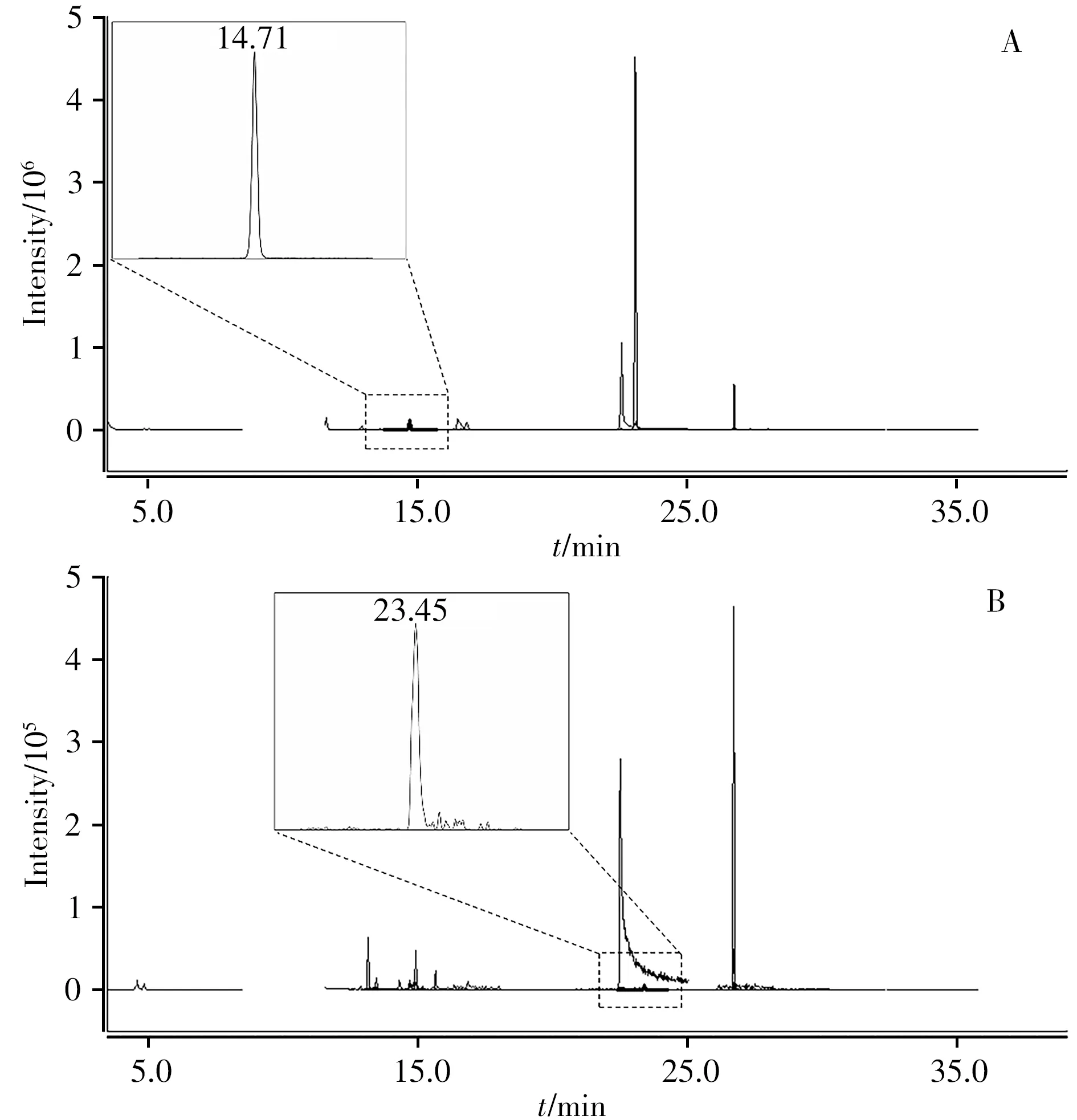
圖2 檢出六氯苯(A)和氟蟲腈(B)的陽性樣品的總離子流圖Fig.2 Total ion chromatograms of hexachlorobenzene(A)and fipronil(B)in positive samples
3 結論
本研究采用乙腈提取,經過冷凍除脂-EMR固相萃取凈化,結合高靈敏度的GC-MS/MS檢測技術,實現了植物源化妝品中53種常見農藥的快速定性定量分析。該方法具有良好的靈敏度、準確度和精密度,前處理簡便快速,能夠滿足實際檢測的要求,為植物源化妝品的質量控制提供了技術支撐。利用本方法在采集的30個植物源化妝品中檢出2個陽性樣品,提示該類化妝品中農藥殘留的問題不容忽視,生產企業與政府相關部門應對該問題予以更高的重視,以促進我國化妝品行業高質量發展。
