PGC-1α調控OPA1改善阿爾茨海默病的機制研究
2022-11-30 04:38:24劉文君史厚珍翟鴻儒張維君朱永強劉子重王玉召王佳
江蘇大學學報(醫學版) 2022年6期
劉文君, 史厚珍, 翟鴻儒, 張維君, 朱永強, 劉子重, 王玉召, 王佳,
(1. 江蘇大學醫學院,江蘇 鎮江 212013; 2. 江蘇大學附屬第四人民醫院神經科,江蘇 鎮江 212001; 3. 濱海縣人民醫院護理部,江蘇 鹽城 224500)
阿爾茨海默病(Alzheimer′s disease, AD)是一種神經退行性疾病,其主要病理學特征是β-淀粉樣蛋白(Amyloid-β, Aβ)沉積,神經纖維纏結以及進行性神經元和突觸丟失[1]。AD的發病機制尚未被完全闡明,膽堿能損傷[2]、神經炎癥[3]、自噬異常[4]、氧化應激[5]、線粒體功能障礙[6]等多種復雜的病理生理過程都與AD的發生有關。近年來研究發現,線粒體功能障礙在AD的發病機制中起著至關重要的作用[7]。AD患者大腦中Aβ的進行性積累可能導致線粒體功能障礙,包括線粒體結構受損、ATP水平降低,線粒體生物合成降低以及線粒體動力學異常[8]。線粒體是一種高度動態的細胞器,處于一種連續不斷的融合和分裂的動態過程,這些持續不斷的運動能有效調節線粒體的形態,控制線粒體在細胞內的分布,以適應細胞生長、分裂、分化過程。線粒體通過快速和可逆的分裂、融合過程,可完成從小圓形細胞器到巨大管狀結構的轉變,位于線粒體外膜的線粒體融合素1(Mitofusin 1, Mfn1)、線粒體融合素2(Mitofusin 2, Mfn2)及線粒體內膜中視神經萎縮癥蛋白1(optic atrophy 1, OPA1)共同協作完成了線粒體的融合[9],已有研究證實線粒體的融合對于其功能的維持至關重要。干擾線粒體的融合,如OPA1的丟失能夠減少線粒體的氧化磷酸化,降低線粒體的膜電勢。在線粒體的融合-分裂過程中,OPA1作為線粒體內膜融合蛋白,對維持線粒體內膜的完整性和線粒體嵴的結構起決定性作用[10]。研究表明,OPA1在線粒體能量維持上也發揮著至關重要的作用[11],增加OPA1水平可以同時增強線粒體融合和ATP的產生[12]。
近年來,越來越多的證據顯示AD的發生與神經元線粒體融合失衡密切相關。AD患者腦組織解剖學研究發現,其海馬神經元線粒體數目明顯減少,并伴隨著線粒體結構破壞,如線粒體嵴斷裂、內部結構丟失及出現短棒狀線粒體[13]。體外研究也已證實轉染了APP質粒的M17細胞出現大量定位于細胞核的碎片化的線粒體,上述線粒體形態及分布的異常伴隨著線粒體融合蛋白表達的改變[14]。
PGC-1α作為過氧化物酶體增殖物激活受體γ(peroxisome proliferator-activated receptor γ, PPARγ)轉錄共激活因子能夠促進線粒體生物合成及抗氧化酶基因的轉錄而被定義為代謝調節靶點[15]。PGC-1α主要存在于高能量需求的組織,如心臟、肝臟、骨骼肌和腦[16]。許多神經生物學疾病的發生都與其腦組織PGC-1α表達下調、抗氧化酶減少及活性氧簇誘導的線粒體功能障礙密切相關[17],如γ-氨基丁酸能神經元敲除PGC-1α能夠模擬精神分裂癥的行為學表型[18]。PGC-1α通過調控基因和環境的相互作用影響了神經突觸的可塑性[18]。帕金森氏綜合征的尸檢報告提示,患者黑質腦區PGC-1α顯著下調,同時伴隨線粒體融合蛋白表達的降低[19]。然而,在AD中PGC-1α究竟扮演了什么樣的角色?PGC-1α是否參與了AD神經元線粒體動力學調控?PGC-1α能否調控OPA1的轉錄及表達,繼而改善AD神經元線粒體的動力學反常?
1 材料與方法
1.1 材料
1.1.1 試劑與抗體 細胞培養基(美國Invitrogen公司),化學試劑(美國Sigma-Aldrich公司),脂質體轉染試劑DNAfection(鎮江愛必夢生物科技有限公司),DAB顯色試劑盒、免疫組化試劑盒(武漢博士德生物工程有限公司),4′, 6-二脒基-2-苯基吲哚二鹽酸鹽(DAPI)、DEPC水(上海碧云天生物技術公司),RNA提取試劑盒(南京諾唯贊生物科技有限公司),cDNA逆轉錄試劑盒(美國Thermo公司)。Aβ抗體(美國CST公司),PGC-1α抗體(北京博奧森生物技術有限公司),OPA1抗體(武漢博士德生物工程有限公司),免疫熒光二抗:Alexa Fluor 594-山羊抗兔IgG(美國CST公司),DyLight 488-山羊抗小鼠IgG(武漢博士德生物工程有限公司)。
1.1.2 質粒和腺相關病毒 APPswe質粒,PGC-1α質粒均由武漢淼靈生物科技有限公司構建。pAAV-MCS-PGC1α-m-FLAG-HA,pAAV-MCS-FLAG-control腺相關病毒購自和元生物技術(上海)有限公司。
1.1.3 細胞系 N2A神經母細胞瘤細胞購自美國ATCC公司。
1.1.4 實驗動物 APP/PS1雙轉基因AD模型小鼠購自美國Jackson實驗室,C57BL/6(WT)小鼠購自江蘇集萃藥康生物科技股份有限公司。
1.2 方法
1.2.1 脂質體轉染 細胞培養至密度為70%時,使用DNAfection進行脂質體轉染,管1:200 μL選擇性培養基(Opti-MEM)+2 μg DNA;管2:200 μL Opti-MEM+5 μL DNAfection;混勻,靜置5 min;管1+管2,混勻,靜置25 min;加入N2A細胞中,次日收集細胞。轉染細胞活力通過細胞免疫熒光或DAPI染色確定。
1.2.2 RNA提取 質粒轉染24 h后,收集細胞,RNA提取試劑盒提取RNA:6孔板每孔加入500 μL裂解液,吹打細胞,轉移至離心管中,反復吹打裂解細胞。向細胞裂解液中加入200 μL DEPC水,混勻后靜置5 min,12 000×g室溫離心、15 min,轉移水相至新的離心管,加入等量異丙醇抽提RNA,混勻后靜置10 min,12 000×g室溫離心、10 min,棄上清液,加入75%乙醇清洗沉淀,8 000×g室溫離心、5 min,棄上清液并晾干,加入20 μL DEPC水溶解沉淀。
1.2.3 RNA逆轉錄為cDNA 向RNA沉淀中加入20 μL DEPC水,測試濃度后,按照cDNA逆轉錄試劑盒說明書將RNA逆轉錄成cDNA。
1.2.4 qRT-PCR檢測APP、PGC-1α和OPA1的mRNA水平 取200 ng cDNA以20 μL體系進行qRT-PCR檢測,以β-肌動蛋白的mRNA作為內參對照,反應條件:95 ℃預變性10 min;95 ℃ 變性15 s,58 ℃ 退火15 s,72 ℃ 延伸30 s,共40個擴增循環。qRT-PCR引物:APP上游5′-TGAATGTGCAGAATGGAAAGTG-3′,下游5′-AACTAGGCAACGGTAAGGAATC-3′;PGC-1α上游5′-GGATATACTTTACGCAGGTCGA-3′,下游5′-CGTCTGAGTTGGTATCTAGGTC-3′;OPA1上游5′-CTTACATGCAGAATCCTAACGC-3′,下游5′-CCAAGTCTGTAACAATACTGCG-3′。
1.2.5 腦區定點微注射AAV-PGC1α 將APP/PS1小鼠麻醉后固定于腦立體定位儀上,使用微量注射器向側頂葉聯合皮層(lateral parietal association cortex, LPtA)(前囟點-1.94 mm,側面±1.5 mm,深度-1.0 mm)注射0.5 μL腺相關病毒(pAAV-MCS-PGC1α-m-FLAG-HA或pAAV-MCS-FLAG-control)。
1.2.6 蘇木素-伊紅染色觀察注射位點 小鼠灌注取腦,經4%多聚甲醛固定過夜后脫水,包埋,切片。石蠟切片脫蠟至水處理,放于蘇木素染液中染色5~10 min,用1%鹽酸乙醇溶液分化數秒,伊紅染液染色1~3 min,染色后的切片脫水透明后,中性樹脂封片。
1.2.7 免疫熒光染色檢測Aβ和PGC-1α陽性細胞數目 小鼠灌注取腦后石蠟切片,石蠟切片二甲苯脫蠟,梯度乙醇復水,將切片置于檸檬酸鹽抗原修復液中微波輻射抗原修復,0.3% TritonX-100穿透細胞30 min,5% BSA+5% 山羊血清封閉90 min,Aβ抗體(1 ∶1 000)和PGC-1α抗體(1 ∶1 000)4 ℃孵育過夜,熒光二抗室溫孵育90 min,DAPI核染10 min,封片。
1.2.8 免疫組織化學染色檢測OPA1陽性細胞數目 小鼠灌注取腦后,石蠟包埋、切片,石蠟切片脫蠟與復水,將切片放入3%過氧化氫溶液中孵育10 min以消除內源性過氧化物酶的干擾,抗原修復、穿透細胞、封閉相關步驟同方法“1.2.7”,4 ℃過夜孵育OPA1抗體(1 ∶1 000)、二抗室溫孵育30 min,滴加DAB顯色,梯度乙醇脫水,中性樹脂封片。
1.2.9 透射電鏡觀察線粒體形態 小鼠灌注取腦后,取腦組織浸于固定液中4 ℃固定24 h,乙醇梯度脫水后包埋。樣品在LEICA EM UC7型超薄切片機中切片,切片經檸檬酸鉛溶液和醋酸雙氧鈾50%乙醇飽和溶液各染色5~10 min,晾干后透射電鏡觀察。
1.3 統計方法
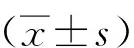
2 結果
2.1 AD小鼠模型及N2A細胞模型PGC-1α表達及轉錄水平降低
以6月齡APP/PS1小鼠作為AD體內模型。通過免疫熒光染色檢測AD模型小鼠LPtA腦區Aβ沉積和PGC-1α蛋白的表達,與WT小鼠相比,APP/PS1小鼠LPtA腦區Aβ沉積顯著增加(t=9.71,P<0.01),驗證了模型的有效性。值得注意的是,AD小鼠LPtA腦區PGC-1α的表達顯著下調(t=9.28,P<0.01)。N2A細胞系脂質體法轉染APPswe構建AD體外模型,qRT-PCR檢測AD組PGC-1α的轉錄水平,與對照組相比,AD模型組APP轉錄水平的增加(t=4.92,P<0.01)伴隨著PGC-1α轉錄水平的下降(t=8.54,P<0.01)。見圖1。
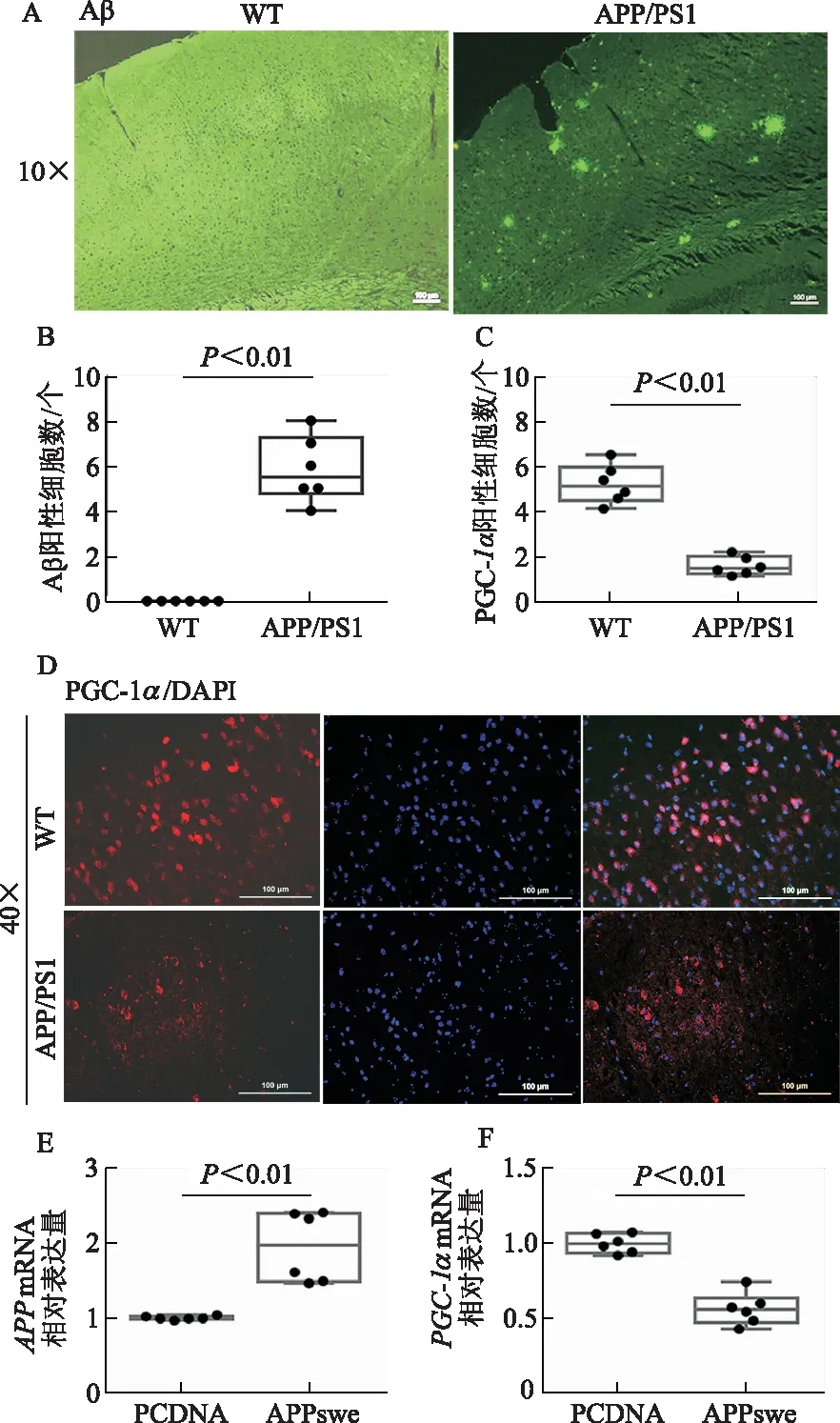
A-B:體內水平評價WT及APP/PS1小鼠LPtA腦區Aβ的表達和定量;C-D:體內水平評價WT及APP/PS1小鼠LPtA腦區PGC-1α的表達和定量;E-F:N2A細胞系脂質體法轉染PCDNA對照質粒或APPswe質粒,體外水平評價對照組及N2A細胞模型組APP mRNA及PGC-1α mRNA的相對表達(圖B的定量面積為1.35 mm2,圖C的定量面積為0.1 mm2,比例尺=100 μm,n=6)
2.2 AD小鼠模型及N2A細胞模型OPA1表達及轉錄水平的降低
以AD模型小鼠為研究對象,通過免疫組化評價OPA1的表達水平,與WT對照組相比,APP/PS1小鼠LPtA腦區OPA1表達降低(t=6.49,P<0.01)。N2A細胞系轉染APPswe構建AD體外細胞模型,qRT-PCR證實mutAPP引起OPA1轉錄水平降低(t=3.28,P<0.01)。見圖2。
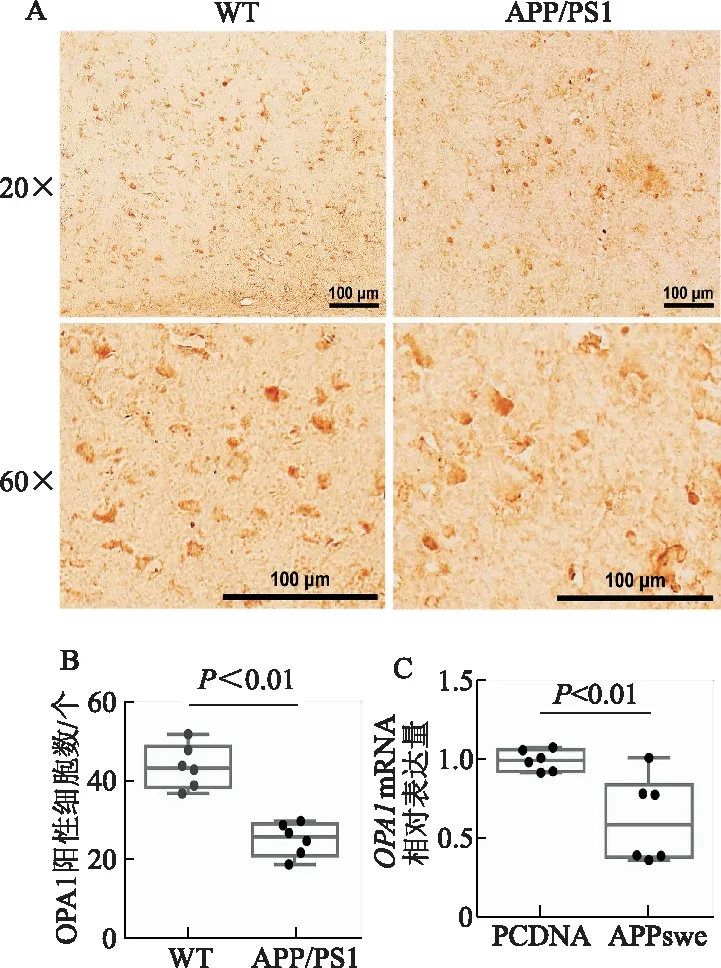
A-B:體內水平評價WT及APP/PS1小鼠LPtA腦區OPA1的免疫組化和定量分析;C:N2A細胞系脂質體法轉染PCDNA質粒或APPswe質粒。體外水平評價對照組及N2A細胞模型組OPA1 mRNA的相對表達(圖B的定量面積為0.03 mm2,比例尺=100 μm,n=6)
2.3 AD模型小鼠線粒體形態異常
透射電鏡觀察顯示,與WT對照組相比,APP/PS1小鼠LPtA腦區神經元線粒體形態受損,內膜損傷的線粒體數目顯著增多(t=6.29,P<0.01)。見圖3。
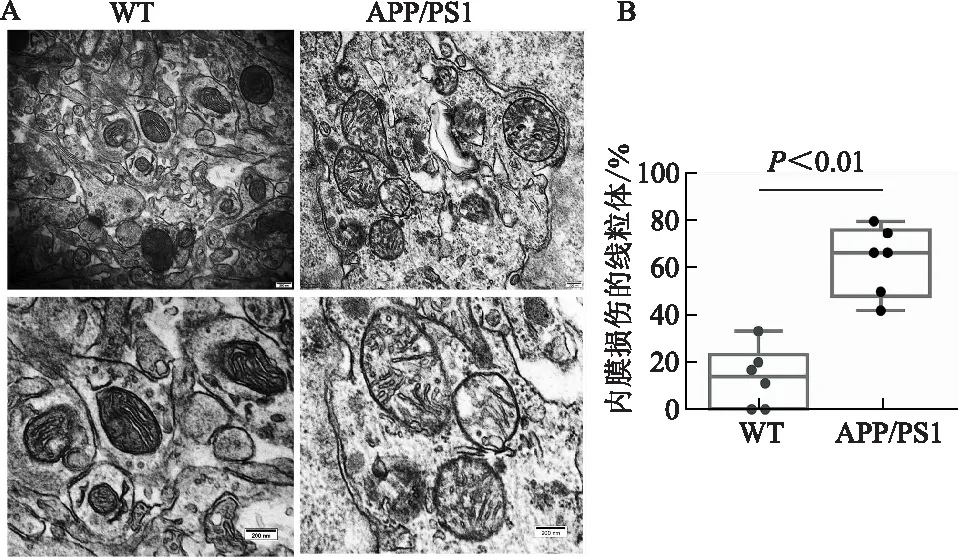
A:體內水平評價WT及APP/PS1小鼠LPtA腦區神經元線粒體形態學改變(透射電鏡);B:內膜損傷的線粒體數量百分比(比例尺=200 nm,n=6)
2.4 PGC-1α抑制了AD時Aβ的轉錄與表達
利用腦區定點微注射及AAV介導的基因轉導技術(pAAV-MCS-PGC1α-m-FLAG-HA),誘導PGC-1α在AD模型鼠LPtA過表達,實驗結束后,HE染色確認灌注位點是否正確,若偏離注射位點,則該樣本的相關數據將被剔除。
利用免疫熒光技術驗證AD模型組及PGC-1α過表達組小鼠LPtA Aβ沉積及PGC-1α的表達情況。與AD模型組相比,PGC-1α在AD模型鼠LPtA成功過表達(t=5.49,P<0.01),且PGC-1α顯著降低了AD模型小鼠LPtA的Aβ沉積(t=4.62,P<0.01)。
N2A細胞系共轉APPswe和PECMV/PGC-1α質粒,qRT-PCR檢測兩組APP和PGC-1α的轉錄水平, PGC-1α轉錄水平明顯增加(t=7.25,P<0.01),與N2A細胞模型組相比,PGC-1α顯著降低了N2A細胞模型組Aβ的轉錄水平(t=9.52,P<0.01)。見圖4。
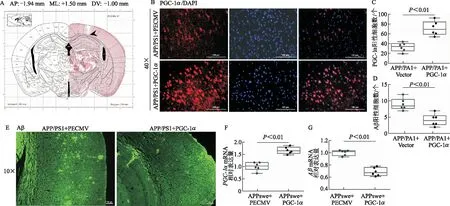
A:小鼠大腦冠狀切片蘇木素-伊紅染色圖;B-C:體內水平確定PGC-1α在APP/PS1小鼠LPtA腦區過表達;D-E:體內水平評價PGC-1α過表達對APP/PS1小鼠LPtA腦區Aβ沉積的影響;F-G:N2A細胞系共轉APPswe和PECMV/PGC-1α質粒,qRT-PCR確認PGC-1α mRNA的相對表達,體外水平評價PGC-1α對Aβ mRNA水平的影響(圖C的定量面積為0.1 mm2,圖D的定量面積為1.35 mm2,比例尺=100 μm,n=6)
2.5 PGC-1α改善了AD發生時OPA1水平的降低
免疫組化驗證PGC-1α過表達對AD小鼠模型LPtA腦區OPA1表達的影響,與對照組(APP/PS1+Vector)相比,PGC-1α過表達顯著增加了AD模型小鼠(APP/PS1+PGC-1α)LPtA腦區OPA1的表達(t=4.09,P<0.01)。N2A細胞系共轉APPswe和PECMV/PGC-1α質粒,qRT-PCR檢測OPA1的轉錄水平,與對照組(APPswe+PECMV)相比,PGC-1α表達顯著增加了N2A細胞模型組(APPswe+PGC-1α)OPA1的轉錄水平(t=4.86,P<0.01)。見圖5。
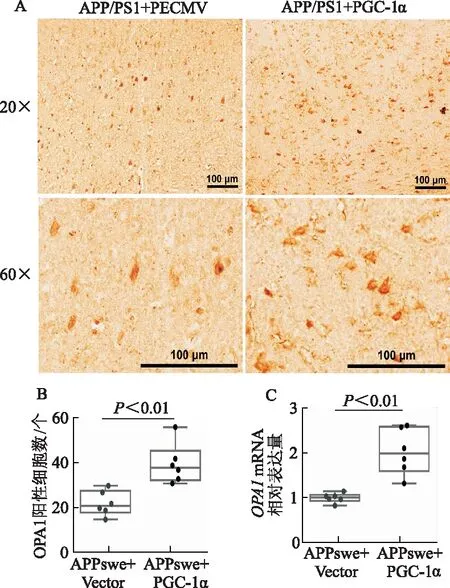
A-B:體內水平評價PGC-1α過表達對APP/PS1小鼠LPtA腦區OPA1表達的影響;C:N2A細胞系共轉APPswe和PECMV/PGC-1α質粒,qRT-PCR評價PGC-1α對OPA1 mRNA水平的影響(圖B的定量面積為0.03 mm2,比例尺=100 μm,n=6)
2.6 PGC-1α改善了AD發生時線粒體的損傷
透射電鏡檢測PGC-1α過表達對AD模型小鼠LPtA腦區神經元線粒體形態的影響,與AD模型組小鼠相比,PGC-1α過表達顯著改善了AD小鼠LPtA神經元線粒體內膜的損傷,線粒體內膜的完整性增加(t=5.72,P<0.01)。見圖6。
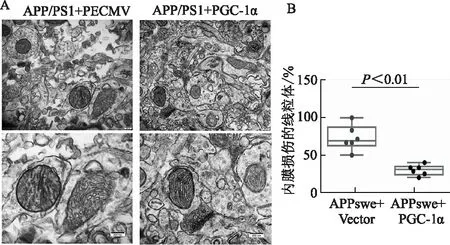
A:體內水平評價PGC-1α過表達對APP/PS1小鼠LPtA腦區神經元線粒體形態的影響(透射電鏡);B:內膜損傷的線粒體百分比的量化(比例尺=200 nm,n=6)
3 討論
近年來,線粒體的動力學改變成為神經生物學領域研究的熱點。線粒體通過快速和可逆的分裂、融合過程,可完成從小圓形細胞器到巨大管狀結構的轉變,OPA1主要定位于線粒體內膜上控制內膜的融合。線粒體內膜的邊界膜和線粒體嵴的交界部分會形成一個結構—嵴連接[20]。線粒體嵴上錨定著大量的與細胞氧化磷酸化和ATP生成相關的重要蛋白,包括呼吸鏈復合物和ATP合成酶。OPA1通過形成聚合體來加強嵴連接點的強度,增加呼吸鏈復合物的穩定性,從而促進線粒體的氧化呼吸效率[21-23]。
越來越多的證據顯示AD的發生與神經元線粒體動力學異常(融合、分裂失衡)密切相關。AD患者腦組織解剖學研究發現,其海馬神經元線粒體數目明顯減少,并伴隨著線粒體結構破壞,如線粒體嵴斷裂、內部結構丟失及出現短棒狀線粒體[8]。體外研究也證實轉染了APP質粒的M17細胞出現大量定位于細胞核的碎片化的線粒體,上述線粒體形態及分布的異常伴隨著線粒體融合及分裂蛋白表達的變化[24]。
PGC-1α作為PPARγ轉錄共激活因子在線粒體生物發生、線粒體質量控制和能量代謝的過程中起到關鍵作用。許多神經生物學疾病的發生都與其腦組織PGC-1α表達下調、抗氧化酶減少及活性氧誘導的線粒體功能障礙密切相關。如帕金森氏綜合征的尸檢報告顯示患者黑質腦區PGC-1α顯著下調,同時伴隨線粒體融合蛋白表達的降低[19]。本課題組前期通過體內實驗已經證實AD的發生伴隨著PGC-1α表達水平的降低及氧化應激產物—DNA損傷標志物8-羥基脫氧鳥苷增加[25]。然而,PGC-1α能否改善AD神經元線粒體動力學異常及其潛在的機制尚不清楚。
本研究通過體內、體外實驗首先評價了AD發生時PGC-1α、OPA1的轉錄及表達水平。結果表明,無論在AD動物模型或APPswe轉染構建的N2A細胞模型中,PGC-1α、OPA1轉錄及表達水平均明顯下降。進一步利用透射電鏡觀察了線粒體形態變化,與Calkins課題組的研究結果一致[8],APP/PS1小鼠的側頂葉聯合皮層神經元線粒體的內膜溶解、破壞,并伴隨線粒體的嵴斷裂。盡管研究證實AD時神經元線粒體形態的改變與PGC-1α及OPA1表達下調有關,然而PGC-1α能否改善AD的發生,是否通過調控線粒體的融合發揮作用還有待進一步探索。
利用6月齡APP/PS1小鼠作為研究對象,通過腦區定點微注射pAAV-MCS-PGC1α-m-FLAG-HA腺相關病毒成功誘導PGC-1α在AD小鼠側頂葉聯合皮層過表達。PGC-1α過表達可以顯著降低AD所伴隨的Aβ沉積。值得注意的是,PGC-1α顯著增加了AD組OPA1的轉錄和表達水平,并且改善了AD所伴隨的線粒體的嵴斷裂和內膜結構。結果表明,PGC-1α很可能通過調控線粒體的內膜融合蛋白OPA1的表達,抑制線粒體形態的損傷,從而達到改善AD的目的。
此外,本研究以N2A細胞系為研究對象,脂質體法共轉APPswe和PECMV/PGC-1α質粒,qRT-PCR評價了PGC-1α過表達對N2A細胞模型中PGC-1α、OPA1及Aβ轉錄水平的調控,與體內結果一致,PGC-1α顯著抑制了APPswe轉染組Aβ的轉錄,該結果伴隨著OPA1轉錄水平的增加。PGC-1α抑制AD神經元線粒體形態的損傷及改善Aβ病理學沉積與其增加OPA1的轉錄及表達水平密切相關。
OPA1的表達經過選擇性剪接和蛋白水解過程,產生多種變體形式,包括線粒體內膜錨定的長鏈OPA1(long OPA1, L-OPA1)和可溶性短鏈OPA1(short OPA1, S-OPA1)。研究發現,L-OPA1不僅可以維持嵴的緊密性,對線粒體融合能力的維持也是不可或缺的[21,26],而S-OPA1是否可以參與線粒體融合,存在爭議[10,21]。有研究證明,S-OPA1在線粒體融合中的作用很小,但可以獨立于L-OPA1維持呼吸功能,在能量維持中發揮作用[27],支持嵴的形成,維持嵴的緊密性[28]。這些研究表明,OPA1對嵴的維持作用獨立于線粒體融合的調控。本研究發現AD的發生伴隨著OPA1表達降低和線粒體嵴的損傷,而PGC-1α可以改善此現象。根據文獻報道,L-OPA1既可以維持嵴的結構,又可以參與線粒體融合,而S-OPA1對線粒體嵴的維持獨立于線粒體融合的作用。PGC-1α調控OPA1的具體機制以及在AD中對L-OPA1和S-OPA1的調控,有待進一步研究。
綜上所述,AD的發生伴隨PGC-1α和OPA1的轉錄及蛋白表達水平下降。PGC-1α通過調控OPA1的表達,改善AD所伴隨的線粒體內膜損傷。PGC-1α很可能為AD的潛在調控位點,當前的研究對AD的機制探索及靶向藥物研發具有重要價值。PGC-1α對線粒體AD融合調控的分子生物學機制有待進一步研究。
猜你喜歡
興趣閱讀·興趣作文與閱讀(低年級)(2025年8期)2025-08-18 00:00:00
美與時代·美術學刊(2022年3期)2022-04-27 01:18:15
今日農業(2021年19期)2022-01-12 06:16:36
中老年保健(2021年11期)2021-08-22 03:15:44
中學生數理化(高中版.高考數學)(2021年1期)2021-03-19 08:28:38
學苑創造·A版(2020年9期)2020-10-13 09:41:02
現代出版(2020年3期)2020-06-20 07:10:34
人大建設(2019年12期)2019-05-21 02:55:32
云南中醫學院學報(2014年3期)2014-07-31 18:57:34
七彩語文·畫刊(2012年3期)2012-04-29 00:00:00
