LC-MS/MS法測定苯磺酸氨氯地平膠囊中兩種潛在基因毒性雜質的含量
2022-12-09 05:18:30孫朗石明睿熊志立
醫藥導報 2022年12期
孫朗,石明睿,熊志立
(1.沈陽藥科大學藥學院,沈陽 110016;2.麗珠集團麗珠制藥廠,珠海 519045)
近年國內仿制藥及一致性評價審評中日益重視雜質譜的研究,特別是潛在基因毒性雜質的評估[1-2]。苯磺酸氨氯地平膠囊臨床應用廣泛,是治療高血壓病的首選藥物之一[3]。由于苯磺酸在合成過程中可能引入苯磺酸甲酯、苯磺酸乙酯、苯磺酸異丙酯等基因毒性雜質,目前國內外對苯磺酸氨氯地平各劑型藥物基因毒性雜質的報道多為配體苯磺酸所引入的苯磺酸酯類雜質[4-5]。作者依據國家藥品監督管理局藥品審評中心“關于公開征求4個ICH指導原則中文翻譯稿意見的通知”和“Q3C(R8):雜質:殘留溶劑的指導原則”,對該品種的起始物料、中間體、副反應及降解產物進行系統研究,篩選出2個分別具有氮雜環氮氧和酰化芳香氨警示結構[6-8]的非苯磺酸酯類潛在基因毒性雜質:氨氯地平氮氧化物和4-(2-(1,3-二氫 -二氧代異吲哚啉-2-基)乙氧基)-3-氧代丁酸乙酯(以下簡稱雜質A和B,結構見圖1),并依據化合物的極性和結構特點,結合所需控制的雜質限度,開發靈敏度高、專屬性強的LC/MS方法[9-11]對其含量進行測定,為苯磺酸氨氯地平膠囊的質量控制提供參考依據。
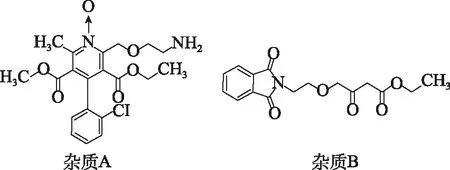
圖1 苯磺酸氨氯地平中潛在基因毒性雜質結構 Fig.1 Potential genotoxic impurities in amlodipine besylate
1 儀器與試藥
1.1儀器 Triple Quad 5500液相色譜-三重四級桿串聯質譜儀(美國 AB Sciex公司);SQP電子分析天平(美國賽多利斯科學儀器公司);KH-500E超聲波清洗器(昆山禾創超聲儀器);H1850離心機(湖南湘儀儀器實驗室開發公司)。
1.2試藥 氨氯地平氮氧化合物(Standardpharm公司,批號:1919336A-SL-01)、4-(2-(1,3-二氫 -二氧代異吲哚啉-2-基)乙氧基)-3-氧代丁酸乙酯(Standardpharm公司,批號:1819345A-SL-01),苯磺酸氨氯地平膠囊(麗珠集團麗珠制藥廠,批號:V180901、V180902、V180903),參比制劑苯磺酸氨氯地平膠囊(Pfizer SA公司,批號:B217802B)。
2 方法與結果
2.1色譜條件 色譜柱為Agilent poroshell 120 EC-C18(4.6 mm×150 mm,2.7 μm);流速為 0.5 mL·min-1;柱溫為40 ℃;自動進樣器溫度5 ℃;進樣體積為2 μL;流動相A為5 mmol·L-1乙酸銨的0.1%甲酸溶液,流動相B為0.1%甲酸乙腈溶液,按以下條件(表1)進行梯度洗脫。

表1 梯度洗脫條件 Tab.1 Conditions of gradient elution
電噴霧離子源(ESI),正離子模式采集,多反應離子監測模式(MRM);雜質A和B的定量離子對分別為 423.0/334.0和320.1/174.0;干燥氣離子源溫度為 600 ℃;干燥氣流速為 35.0 psi;霧化離子化電壓為 5500 V;霧化氣50 psi;輔助加熱氣50 psi;傳輸電壓為 85 V;碎裂電壓為 15 V;掃描范圍為m/z100~500,質譜圖見圖2。
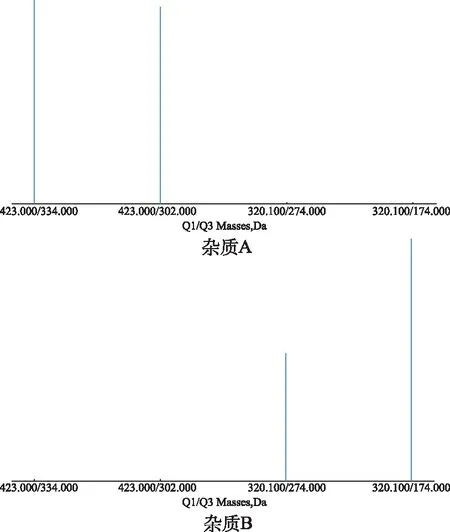
圖2 雜質A和B的質譜圖 Fig.2 Mass spectrum of impurity A and B
2.2溶液制備
2.2.1對照品溶液的制備 精密稱取雜質A和B對照品適量,用乙腈稀釋成每毫升含雜質A 22 ng·mL-1、雜質B 22 ng·mL-1的溶液,作為對照品溶液。
2.2.2供試品溶液的制備 精密稱取苯磺酸氨氯地平膠囊內容物適量(相當于苯磺酸氨氯地平5.0 mg),置50 mL離心管中,精密加入溶劑乙腈25 mL,渦旋,超聲5 min,10 000 r·min-1離心5 min,取上清液作為供試品溶液。
2.2.3空白溶液的制備 取乙腈濾過后即可。
2.3專屬性考察 取各個雜質的對照品溶液、加標供試品溶液(100%濃度水平)及空白溶液,依法測定。得到的色譜圖表明:各雜質對照品溶液及加標供試品溶液在同一位置顯示目標分子量峰,空白溶液無干擾,本方法專屬性良好,典型色譜圖見圖3。
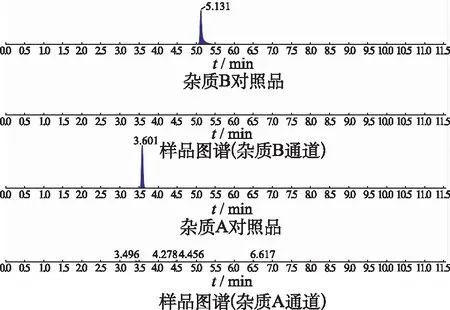
圖3 雜質對照品及樣品質譜圖 Fig.3 Mass spectrum of impurities and sample
2.4定量限與檢測限考察 精密稱取各雜質對照品適量,加乙腈配制成每毫升含雜質A約4.5 ng·mL-1,雜質B約4.3 ng·mL-1的溶液,作為定量限溶液,依法檢測6次。結果表明:各雜質6次測試結果信噪比約等于10;雜質A和B連續6針定量限溶液中目標峰峰面積的RSD分別為2.0%和0.8%,本方法定量限能滿足需求。
精密量取定量限溶液2.5 mL,置于5 mL量瓶中,加乙腈定容,制成每毫升含雜質A約2.3 ng·mL-1,雜質B約2.2 ng·mL-1的溶液,作為檢測限溶液,依法檢測6次。結果表明:各雜質6次測試結果信噪比約等于3,本方法檢測限能滿足需求。
2.5線性關系考察 精密稱取雜質A和B對照品各10.255和9.986 mg,分別置于100 mL量瓶,加乙腈溶解并定容至刻度。精密量取上述溶液各0.22 mL置于100 mL量瓶,加乙腈定容至刻度,制得含兩雜質濃度分別為225.6和219.7 ng·mL-1的雜質對照品儲備液(1000%限度濃度)。分別精密量取上述對照品儲備液0.2,0.5,1,1.5和2,0 mL置于10 mL量瓶,加乙腈定容至刻度,制得含雜質A濃度為4.51,11.28,22.56,33.84,45.12 ng·mL-1;雜質B濃度為4.39,10.98,21.97,32.95,43.93 ng·mL-1各共5個濃度水平的線性溶液。將上述溶液依次依法測定,結果見表2。以濃度為橫坐標,峰面積為縱坐標,進行線性回歸,可得雜質A線性方程為Y=8.164×104X+70 146(r=0.990);雜質B線性方程為Y=2.362×105X+35 610(r=1.000)。結果表明:雜質A在4.51~45.12 ng·mL-1,雜質B在4.39~43.93 ng·mL-1濃度范圍內,線性關系良好。

表2 線性考察實驗結果 Tab.2 Linearity of two kinds of impurities
2.6精密度實驗 取“2.5”項下各雜質100%濃度水平溶液(含雜質A和B的濃度分別為22.56和21.97 ng·mL-1),依法連續測定6次,分別計算雜質A和B含量的RSD值。結果表明:各雜質6次測定結果的RSD分別為0.6%和0.5%,本方法精密度良好。
2.7重復性實驗 另由不同研究實驗人員在不同時間重復“2.6”項下實驗,重新配制各雜質100%濃度水平溶液(含雜質A和B的濃度分別為22.52 ng·mL-1和22.03 ng·mL-1),依法連續測定6次,分別計算雜質A和B含量的RSD值。結果表明:各雜質6次測定結果的RSD分別為0.6%和0.8%,二名研究實驗人員共12次測定中,雜質A的RSD為1.6%,雜質B的RSD為2.2%。本方法中間精密度良好。
2.8穩定性實驗 取“2.5”項下雜質對照品儲備液(1000%限度濃度)10 mL置于100 mL容量瓶,加乙腈定容至刻度,作為對照品溶液。精密稱取苯磺酸氨氯地平膠囊內容物適量,置50 mL離心管中,加入25 mL對照品溶液,渦旋,超聲5 min,10 000 r·min-1離心5 min,取上清液作為加標供試品溶液。在0,12,25 h依法檢測,并與初始測定結果進行對比。結果表明:對照品溶液和加標供試品溶液放置25 h后,各個雜質含量的變化均在80~120%之間,對照品溶液和供試品溶液在25 h內均穩定。
2.9加樣回收率實驗 配制“2.5”項下雜質對照品儲備液,分別精密量取2.5,5和7.5 mL至50 mL容量瓶,加乙腈定容至刻度,制得50%,100%和150%濃度對照品溶液。精密稱取苯磺酸氨氯地平膠囊內容物適量(相當于苯磺酸氨氯地平5.0 mg),分別置于3個50 mL離心管中,分別精密量取加入上述3個濃度對照品溶液25 mL,旋渦,超聲5 min,10 000 r·min-1離心5 min,取上清液,作為50%、100%、150%濃度加標供試品溶液。各濃度加標供試品溶液平行配置3份。取上述溶液依法檢測,計算各濃度平均回收率,并計算RSD值,結果見表3,4。
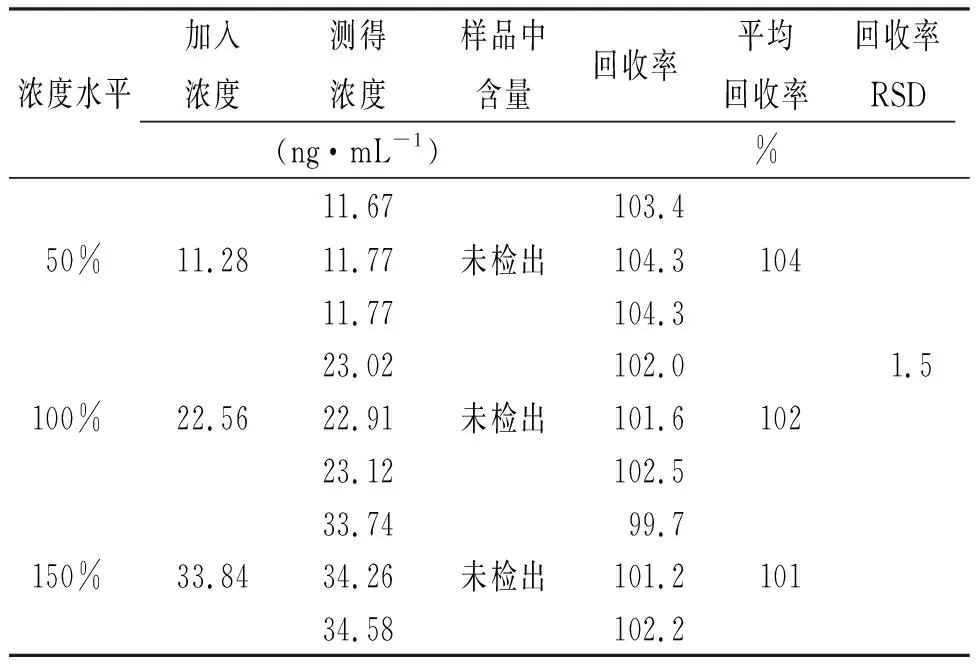
表3 雜質A加樣回收率的測定結果 Tab.3 Recovery result of impurity A
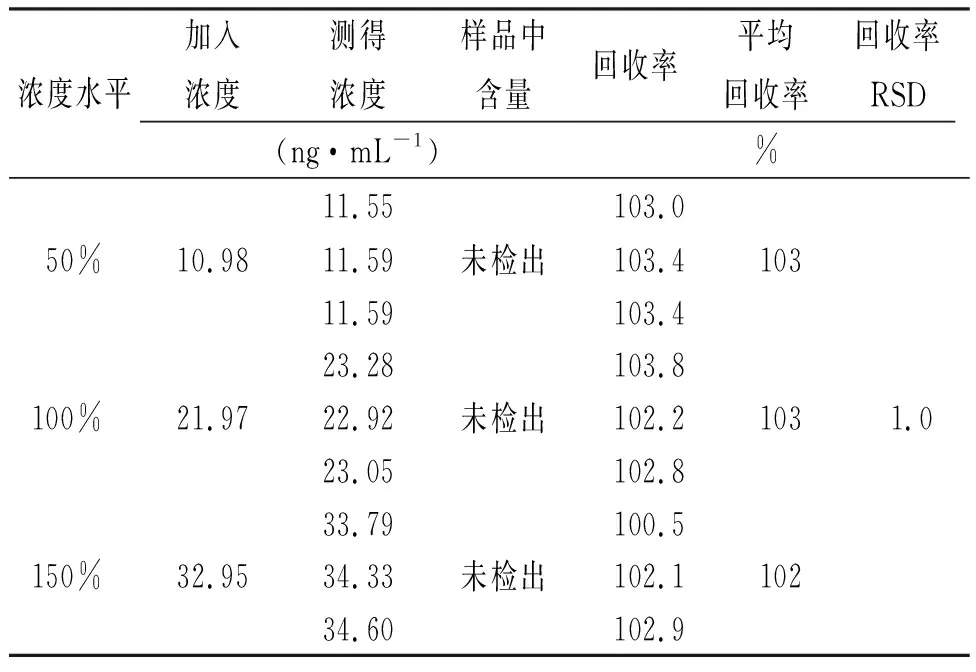
表4 雜質B加樣回收率的測定結果 Tab.4 Recovery result of impurity B
結果表明:雜質A加樣回收率范圍為99.7%~104.3%,RSD為1.5%;雜質B加樣回收率范圍為100.5%~103.8%,RSD為1.0%。本方法準確度良好。
2.10樣品含量測定 取麗珠集團麗珠制藥廠不同批號的樣品3批(批號:V180901,V180902,V180903),Pfizer SA參比制劑1批(批號:B217802B),按“2.2.2”項下方法制備供試品溶液,依“2.1”項下方法測定,計算出不同樣品中雜質A和B的含量。結果上述4批樣品中均未檢測出雜質A和B。
3 討論
依據ICH M7和《中華人民共和國藥典》2020年版“遺傳毒性雜質控制指導原則”[12],結合苯磺酸氨氯地平膠囊的最大日用劑量,使用毒理學關注閾值(TTC)計算出苯磺酸氨氯地平膠囊中兩種潛在基因毒性雜質的可接限度為110×10-6。由于基因毒性雜質在極低的濃度水平時也可能導致DNA突變,其測定分析有較強的挑戰。待測定的潛在基因毒性雜質從結構上看均含有苯環基團,有一定的紫外吸收強度,但預實驗結果表明,HPLC紫外檢測器靈敏度無法滿足雜質控制的需求,故選用靈敏度高、專屬性強的LC-MS/MS方法,對其含量進行測定。
實驗中考察了不同類型的色譜柱,結果表明Agilent poroshell 120 EC-C18(4.6 mm×150 mm,2.7 μm)上的分離度和峰型最佳。由于該色譜柱填料粒度較細,為2.7 μm,為避免壓力過大,優選流速為0.5 mL·min-1;流動相A和B中均加入0.1%甲酸,用以提高待測定組分在質譜中的離子化效率,改善其峰型。
從兩個潛在基因毒性雜質的來源上來看,雜質B為氨氯地平合成過程中的中間體,為非降解雜質,僅有較低的殘留風險,建議在原料藥質量標準中進行控制;雜質A為氨氯地平的潛在氧化降解雜質,應結合多批次穩定性數據評估風險。
從檢測結果來看,國產與原研進口的苯磺酸氨氯地平膠囊均未檢出上述潛在基因毒性雜質,質量趨向一致。本研究首次開發LC-MS/MS法對苯磺酸氨氯地平膠囊中的非磺酸酯類潛在基因毒性雜質進行含量測定,所開發的方法也可供苯磺酸氨氯地平原料藥及其他劑型參考,為進一步規范強化本類藥物的質量提供技術支持。
