基于固相萃取-響應面優化的碳骨架在線催化/GC-MS/MS技術測定含油金屬原料表面痕量氯污染物
2023-01-05 11:16:26張子豪鄭建國麥曉霞彭速標劉瑩峰
分析測試學報 2022年12期
張子豪,鄭建國,麥曉霞,肖 前,彭速標,劉瑩峰,李 丹
(廣州海關技術中心,廣東 廣州 510623)
長期以來,我國金屬冶煉行業面臨節能減排的環保壓力和高品質原料不足的問題,從國外進口相對短缺的金屬廢物原料成為解決我國金屬資源不足的有效方案。2018年到2021年,我國數次調整《進口廢物管理目錄》,進口金屬廢物原料陸續被列入《禁止進口固體廢物目錄》[1]。由于國內金屬冶煉產業對原料的需求依然旺盛,進口國外金屬資源可以滿足國內的部分原料需求,因此從國外進口高品質再生金屬原料成為新趨勢。但進口金屬再生料可能存在品質參差不齊、夾雜物超標、有害物質超標等環保風險,其中粉塵、污泥、纖維等夾雜物易被識別與檢測,而含油金屬再生料中的油污特別是油污中含氯污染物因基質干擾大、目標物異構體多、含量較低等原因而難以準確測定。油污中含氯污染物(氯化石蠟、多氯聯苯、多氯萘等物質)的普遍存在使其受到行業越來越多的關注。此類物質也是監管部門的重點關注對象。我國于2019年至2020年發布實施了GB/T 39733-2020《再生鋼鐵原料》、GB/T 38472-2019《再生鑄造鋁合金原料》、GB/T 38471-2019《再生銅原料》等系列再生金屬標準[2-4]。其中規定再生金屬原料中危險廢物的質量不應超過總質量的0.01%。
再生金屬原料主要由廢舊金屬通過簡單拆解切割、篩分、沖洗后獲得,可能會殘留原設備工作環境或處理過程中加入的絕緣油、液壓油、切削油等。其中可能含有亞老哥爾(工業多氯聯苯)、鹵蠟(工業多氯萘)、氯化石蠟(短鏈和中鏈)等耐熱、阻燃、增塑類物質[5]。目前國內外對多氯聯苯、多氯萘、氯化石蠟等物質的常規檢測技術均有一些報道,如采用氣相色譜-電子捕獲檢測器測定食品包裝材料和皮革中的短鏈氯化石蠟[6-7],氣相色譜-高分辨質譜法測定生物環境樣品中的多氯聯苯與多氯萘[8-9],氣相色譜-質譜法測定玩具及泥土樣品中的短鏈氯化石蠟與多氯聯苯[10-12],液相色譜或質譜法測定食品和塑料等樣品中的氯化石蠟、多氯聯苯等[13-16],然而針對再生金屬原料中含氯污染物的測定和在線催化技術的應用未見報道。含氯有機污染物為人工合成物質,主要由對應的芳烴與脂肪族正構烷烴氯化制得,由于碳骨架氯化位置及氯化度等均存在差異,使有機氯化物存在上百種異構體[17]。實際測定過程中目標物結構復雜,缺少對應的標準品,多氯聯苯混合物特別是氯化石蠟混合物的分離效果不理想,峰形均為寬譜帶的峰簇,當目標物共同存在時峰形均有一定疊加,特征離子也可能部分重疊而難以識別,導致靈敏度下降等均是此類物質難以準確定量的重要原因。
本研究以超聲浸漬法合成的Pd/改性活性炭為催化劑,將目標物在富氫環境下在線脫氯加氫,含氯污染物轉化為特征直鏈烷烴和芳烴,使測定目標從多氯烴分散的同位素離子轉換為集中的高響應烷烴、芳烴離子,進一步減少干擾,提高檢出限,并利用氣相色譜-串聯質譜多反應監測模式測定。該方法靈敏度高、準確穩定,適用于金屬原料中多氯聯苯(PCBs)、多氯萘(PCNs)、短鏈和中鏈氯化石蠟(SCCPs、MSCCPs)等含氯物質的快速篩查和測定。
1 實驗部分
1.1 儀器與試劑
Agilent 7890B-7000C氣相色譜-質譜/質譜儀(美國Agilent公司);Precision Hydrogen 100氫氣發生器(英國Peak公司);AS20500A超聲波發生器(加拿大AUTO Science公司);可調移液器(1~100 μL,德國普蘭德公司);DB-5MS UI石英毛細管柱(30 m×0.25 mm,0.25 μm,美國Agilent公司);不分流單錐襯管(容量900 μL,內徑4 mm,美國Agilent公司);脫活玻璃棉(色譜級,美國Agilent公司);活性炭粉(200目,上海麥克林生化科技有限公司);自制固相萃取柱(硝酸銀硅膠固相萃取柱:1 g/6 mL)[18]。
C10~C17正構烷烴(正癸烷、正十一烷、正十二烷、正十三烷、正十四烷、正十五烷、正十六烷和正十七烷)、四氫萘(KDTN)、苯基環己烷(HCB)、正戊基苯、一氯代烴(1-氯癸烷、1-氯十一烷、1-氯十二烷、1-氯十三烷、1-氯十四烷、1-氯十六烷、1-氯萘和2-氯聯苯)、1,1,1,3,11,13,13,13-八氯十三烷、八氯萘、2,2,3,4,5-五氯聯苯、2,2,3,3,4,4,5,5,6-九氯聯苯、短鏈氯化石蠟(100 μg/mL,平均氯化度51.5%)、中鏈氯化石蠟(100 μg/mL,平均氯化度52.0%)、多氯萘1014(100 μg/mL,平均氯化度63.5%)和多氯聯苯1260(100 μg/mL,平均氯化度60.0%)標準物質均購自德國Dr.Ehrenstorfer GmbH公司,純度≥98%。正己烷、二氯甲烷(HPLC級,美國Tedia公司);無水乙醚(分析純,99%,廣州化學試劑廠);氯化鈀(99.9%,廣州市丹安儀器儀表有限公司);冰醋酸(99.5%,廣州化學試劑廠);氨水(分析純,廣州化學試劑廠)。
1.2 標準工作溶液的配制
稱量一定質量的上述還原產物(C10~C17正構烷烴、四氫萘、苯基環己烷)和一氯代烴(1-氯癸烷、1-氯十一烷、1-氯十二烷、1-氯十三烷、1-氯十四烷、1-氯十六烷、1-氯萘和2-氯聯苯)共18個標準物質于18個10 mL棕色容量瓶中,用正己烷稀釋定容,配制質量濃度均為1 000 μg/mL的還原產物標準儲備液和一氯代烴標準儲備液。內標儲備液:稱量一定量的正戊基苯于10 mL棕色容量瓶中,用正己烷配制1 000 μg/mL的內標標準儲備液。工作溶液:使用還原產物標準儲備液和內標儲備液,用正己烷配制目標物質量濃度均為0.02、0.05、0.10、0.25、0.50、1.00 μg/mL的還原產物標準工作溶液,內標質量濃度為0.25 μg/mL。使用一氯代烴標準儲備液和內標儲備液,配制質量濃度為1.00 μg/mL的一氯代烴標準工作溶液,內標質量濃度為0.25 μg/mL。催化效率質控溶液:使用短鏈氯化石蠟(氯化度為51.5%)、中鏈氯化石蠟(氯化度為52.0%)、多氯萘1014和多氯聯苯1260標準溶液配制各物質質量濃度均為0.50 μg/mL的質控溶液,內標質量濃度為0.25 μg/mL。
1.3 樣品預處理
稱取50.0 g(精確至0.01 g)金屬原料碎料樣品于250 mL旋蓋瓶中,加入150 mL正己烷,40℃下超聲萃取30 min,溶液轉移至150 mL平底燒瓶,旋蒸濃縮至15 mL后轉移至25 mL容量瓶中,正己烷定容。移取以上溶液1 mL至已用0.5 mL正己烷活化的硝酸銀硅膠固相萃取柱中,待充分吸收后加入3.0 mL正己烷-乙醚(體積比9∶1)混合溶液,當溶液全部通過萃取柱篩板后,棄去以上淋洗液,更換接收瓶。繼續向固相萃取柱中加入2.5 mL正己烷-乙醚(1∶1)混合溶液,收集上述洗脫液,氮吹至干后,用配有內標(質量濃度為0.25 μg/mL)的正己烷溶液定容至1 mL,過濾后待測定。
1.4 催化劑的制備和反應襯管的填裝
稱取10 g活性炭加入150 mL煮沸后的10%硝酸水溶液中,攪拌處理30 min,用去離子水多次沖洗至中性后過濾,得到的活性炭用去離子水超聲10 min,過濾并在60℃下干燥得到改性活性炭。
采用超聲輔助浸漬法制備Pd/改性活性炭催化劑前驅體[7],稱取84 mg固體氯化鈀粉末于50 mL燒杯中,加入20 mL 0.06 mol/L鹽酸溶液,45℃攪拌溶解,加入5 g改性活性炭和1.5 g聚乙二醇分散,超聲20 min后靜置。將上述溶液置于95℃干燥箱中干燥得到黑色粉末,即為Pd/改性活性炭催化劑前驅體。
取不分流單錐襯管,底部填入脫活玻璃棉后,從下往上依次加入1 mm高度的碳酸鈣和10 mm高度的Pd/改性活性炭催化劑前驅體,最后覆蓋一層玻璃棉,將該襯管裝入氣相色譜進樣口,320℃下以氫氣為載氣活化1 h,即得Pd/改性活性炭催化劑反應襯管。
1.5 氣相色譜-串聯質譜條件
色譜柱:DB-5MS UI石英毛細管柱(30 m×0.25 mm,0.25 μm);載氣:高純氫氣;碰撞氣:高純氮氣;柱流速:1.5 mL/min;進樣口溫度:260℃;分流比:不分流;起始溫度:45℃(保持3 min),以15℃/min升至250℃(保持1 min),最后以30℃/min快速升至300℃。
電子轟擊離子源(EI),能量:70 eV;傳輸線溫度:300℃;離子源溫度:230℃;四極桿溫度:150℃;多反應監測(MRM)負離子模式,溶劑延遲5 min。
2 結果與討論
2.1 凈化與洗脫條件的優化
非極性溶劑可將樣品洗出液中的非極性雜質(如礦物油、切削油中脂肪族烴等物質)去除,實驗采用硝酸銀硅膠固相萃取柱作為凈化柱,分別以環己烷、正己烷、正戊烷、二氯甲烷4種溶劑作為淋洗溶劑。配制含有正十二烷、正十四烷、四氫萘、苯基環己烷、短鏈氯化石蠟(氯化度51.5%)和多氯聯苯1260質量濃度均為1.00 μg/mL的混合標準溶液,分別用1.0、1.5、2.0、2.5、3.0 mL的淋洗溶劑對上述標準溶液進行回收率實驗。結果表明,以相同體積正己烷或正戊烷淋洗時,正構烷烴的回收率較高,四氫萘和苯基環己烷的回收率較低;環己烷與正己烷的淋洗效果相似,四氫萘和苯基環己烷的回收率略有改善;二氯甲烷由于極性較大,易將正構烷烴、四氫萘、苯基環己烷與少量氯化石蠟和多氯聯苯一起洗出,因此單一溶液均不適合作為淋洗溶劑。選用正己烷-乙醚(9∶1)和正己烷-二氯甲烷(9∶1)混合溶劑分別作為凈化淋洗溶劑進行實驗,結果表明,正己烷-二氯甲烷(9∶1)的洗脫能力稍強。這是由于二氯甲烷相對于乙醚的溶解范圍較大,溶解性好。但當苯基環己烷完全洗出時伴有少量氯化石蠟被同步帶出,因此選擇正己烷-乙醚(9∶1)混合溶液作為淋洗溶劑。考察了不同體積淋洗溶劑對目標化合物回收率的影響,使用2.0 mL淋洗溶劑時,正構烷烴已完全洗出;3.0 mL淋洗溶劑時,四氫萘和苯基環己烷的回收率達到96%以上;淋洗溶劑達3.5 mL以上時有少量短鏈氯化石蠟和多氯聯苯被洗出,因此選擇3.0 mL正己烷-乙醚(9∶1)混合溶劑作為淋洗溶劑。
硝酸銀硅膠柱分離飽和烴、芳烴類與氯代化合物的效果較好,這可能歸因于銀離子與目標物氯原子形成的弱絡合作用,從而實現了對含氯烴較強的保留。樣品經正己烷-乙醚(9∶1)淋洗凈化后,萃取柱上的含氯化合物需用合適的洗脫溶劑洗脫出來。氯化烷烴和芳烴類物質因碳骨架上氯化位置不同而具有一定極性,可被二氯甲烷、酯類、醚類等溶劑洗脫,因此選擇極性不同的二氯甲烷-乙醇(5∶1)、二氯甲烷、正己烷-二氯甲烷(1∶1)、正己烷-乙醚(1∶1)、正己烷-乙酸乙酯(1∶1)作為洗脫溶劑。選擇不同體積(1.5、2.0、2.5、3.0、3.5 mL)的洗脫溶劑,用上述一氯代烴標準工作溶液和催化效率質控溶液(混合氯代烴)進行回收率實驗,每個條件進行2次實驗。結果表明,加入相同體積的洗脫溶劑時,前2種溶劑的洗脫效果均較理想,回收率達95%以上,但溶劑極性較強,洗脫出樣品中芳烴類和酯類物質,可能會對芳烴和直鏈烷烴類還原產物造成干擾。正己烷-乙酸乙酯混合溶劑的洗脫效果一般,平均回收率約在85%左右;正己烷-乙醚和正己烷-二氯甲烷混合溶劑對氯化石蠟和多氯聯苯類物質的洗脫效果均較好,洗脫體積在2.5 mL以上時回收率達到95%,但混合溶劑中含有過多的二氯甲烷可能會影響后續催化反應的效率。綜合考慮,采用2.5 mL正己烷-乙醚(1∶1)混合溶液作為洗脫溶劑。
2.2 催化劑的選擇
分別采用水浴浸漬法制備Pd/玻璃珠載體催化劑[7]、超聲輔助浸漬法制備Pd/改性活性炭催化劑、Pt-Sn/氧化鋁催化劑[19]和Pt-Ag/活性炭催化劑[20]。以上4種催化劑均在320℃下用氫氣活化1 h,活化后保持干燥。采用填有上述4種催化劑的反應襯管,對1-氯十三烷和八氯十三烷標準溶液進行脫氯加氫催化反應,對比脫氯催化效果。實驗表明,上述2種氯代烴在4種不同的催化襯管中均能轉化為對應的還原產物。雙金屬催化體系中,Pd-Ag/活性炭催化劑因Ag的加入催化活性有所降低,還原產物的轉化率較低,高氯化合物產物中還含有部分未充分還原的低氯化合物,如三氯十三烷。Pt-Sn/氧化鋁催化劑的催化活性較高,能對烯烴和酮類物質產生加成反應,增加了樣品分析的復雜性。Pd/玻璃珠載體催化劑和Pd/改性活性炭催化劑的催化效果適中,其中以改性活性炭為載體的催化效率更高,這可能與活性炭經硝酸預處理和超聲輔助浸漬處理有關。硝酸的氧化作用使活性炭表面的氧官能團增加而增強了其吸附能力,加之超聲輔助也能更好提高活性炭載體的吸附性能,使活性金屬Pd能均勻地分布在活性炭的內外表面,因此催化效率更高。最終選擇Pd/改性活性炭催化劑作為脫氯加氫催化劑。圖1為氫氣活化后的Pd/改性活性炭催化劑與Pd/改性活性炭催化劑前驅體的二維X射線衍射譜圖對比,活化后催化劑在2θ=40°左右處有1個較小的鈀衍射峰,說明還原產生了金屬鈀,并附著于改性活性炭上。
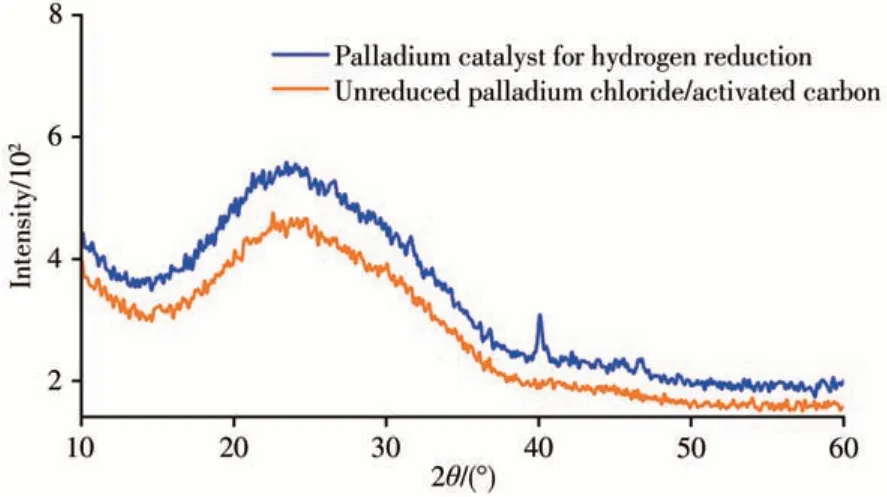
圖1 Pd/改性活性炭催化劑的二維X射線衍射譜圖Fig.1 Two-dimensional X-ray diffraction patterns of Pd/activated carbon catalyst
2.3 硝酸濃度對催化活性的影響
本實驗發現,經硝酸改性的活性炭載體鈀催化劑的催化活性優于未經硝酸改性的活性炭載體鈀催化劑。這是因為經硝酸氧化改性后,活性炭表面的含氧官能團數量顯著增加,含氧官能團通過氫鍵、共價鍵和其他物質相互作用,可提高活性炭的吸附能力。實驗考察了質量分數分別為5%、10%、15%、20%、25%的硝酸水溶液對活性炭改性后催化活性的影響。結果表明,用10%硝酸改性處理的活性炭催化效果最好,硝酸濃度大于10%后,催化活性逐漸降低。這可能是由于高濃度的硝酸改性處理活性炭時降低了載體的比表面積,導致催化活性下降,最終選擇10%硝酸為活性炭改性用硝酸的濃度。
2.4 催化襯管條件的優化
2.4.1 溫度對催化效率的影響反應襯管中催化劑在氫氣環境中活化,以一氯代烴標準工作溶液為研究對象,考察了進樣口溫度(200、220、240、260、280、290、300、310℃)對催化效率的影響,以進樣口溫度對各一氯代烴還原產物(C10~C14及C16飽和直鏈烴、苯基環己烷和四氫萘)的色譜響應作圖(如圖2所示)。結果表明,8種一氯代烴在不同溫度下還原,氯代脂肪烴產物的響應峰面積隨溫度升高而升高,在240~280℃左右峰面積最高,溫度高于290℃,峰面積有所下降。氯代芳烴的還原現象與氯代脂肪烴有所區別,氯代芳烴隨催化溫度的變化會生成不同的還原產物。如多氯聯苯與多氯萘的還原產物較復雜,多氯聯苯、多氯萘除了能夠脫氯加氫轉變為聯苯和萘,還會進一步還原生成苯基環己烷、四氫萘和少量甘菊環烴類物質,多氯萘和多氯聯苯在200~280℃范圍內的主要還原產物為四氫萘和苯基環己烷,還原產物的峰面積與溫度成反比,溫度升高會抑制還原產物四氫萘和苯基環己烷的生成。溫度高于270~280℃左右,還原產物萘和聯苯的含量上升,四氫萘和苯基環己烷的含量下降。

圖2 溫度對催化性能的影響Fig.2 Effect of temperature on catalytic performance
2.4.2 氫氣流速對催化效率的影響載氣作為還原氫的來源使樣品中的氯代烴在氫氣氛圍下催化還原。氫氣流速對催化劑的催化效率有一定影響,通過設置不同的氫氣流速(1.0、1.2、1.4、1.6、1.8 mL/min)對1-氯萘和2-氯聯苯的標準溶液進行測試。結果顯示,氫氣流速從1.0 mL/min升至1.8 mL/min時,還原產物的保留時間逐漸變小,峰面積緩慢變大。這可能是由于在相對較高的氫氣流速下,進樣口壓力有所上升,目標物質與催化劑的接觸更加充分。流速為1.4~1.6 mL/min時,催化效率趨于穩定。
2.4.3 氯化程度對催化效率的影響選取1-氯十三烷(MCTRID)、1,1,1,3,11,13,13,13-八氯十三烷(OCTRID)、1-氯萘(MCN)、八氯萘(OCN)、2-氯聯苯(PCB1)、2,2,3,4,5-五氯聯苯(PCB87)和2,2,3,3,4,4,5,5,6-九氯聯苯(PCB206)儲備液,配制成質量濃度均為10 μg/mL的工作溶液,在“1.5”條件下進行脫氯加氫還原實驗,催化效率如表1所示。結果表明,分子中氯的質量分數越低,還原得到的烴類物質豐度越大,峰面積越大。7種化合物的催化效率均不低于95.1%,可以看出催化劑對一氯代烴和多氯代烴的催化效率無明顯差別,說明氯含量對催化劑的催化效率影響較小。

表1 不同氯含量的一氯代烴和多氯代烴的催化效率Table 1 Catalytic efficiencies of chlorinated hydrocarbons with different chlorine contents
2.4.4 催化參數的優化脂肪族氯代烴和芳香族氯代烴的脫氯規律不同,因此需對催化條件進行優化,以1-氯十三烷和2-氯聯苯為脂肪族氯代烴和芳香族氯代烴的代表物質,采用響應面模型確認最優參數。氯化程度對催化效率的基本影響較小,因此選取催化過程中影響較大的催化溫度(A,℃)、氫氣流速(B,mL/min)和可能對催化效率有影響的化合物質量濃度(C,μg/mL)、催化劑用量(D,g)4個參數作為變量,以某金屬原料加標樣品進行實驗,每一變量設置3個水平,試液中1-氯十三烷和2-氯聯苯的催化效率為響應值,根據Box-Behnken中心組合實驗設計原理,分別進行4因素3水平共29次實驗。得到四因素交互影響顯著的1-氯十三烷和2-氯聯苯的催化效率響應面圖如圖3所示,氯代脂肪烴的回歸模型方程為:catalytic efficiency=88.40+4.42A-0.92B-0.67C+0.50D+1.50AB+0.75AC+1.00AD+1.75BC-0.50BD-12.99A2-1.74B2-0.12C2-2.12D2;氯代芳烴的回歸模型方程為:catalytic efficiency=73.60-14.67A+2.25B-1.00C+1.75D+1.75AB+0.50AC-0.25AD-0.25BC-4.74BD-3.25CD-7.34A2+0.03B2+0.66C2-2.47D2。

圖3 四因素交互影響顯著的1-氯十三烷和2-氯聯苯的催化效率響應面圖Fig.3 Response surface and contour of the recoveries for MCTRID and PCB1 affected alterately by four factors
對上述實驗結果進行方差分析,由表2可知該模型的F值為6.75和19.76,模型概率p值均小于0.05,說明各影響因子對催化效率的影響顯著。通過軟件擬合得到調整相關系數分別為0.871 0和0.951 8,模型擬合度良好,說明相關性較好,能解析模型87.10%和95.18%的響應值變化,可以此模型對再生金屬原料中氯代烴的催化效率進行分析。由兩個模型的F值可知該模型一次項A的影響均極顯著,C的影響不顯著。單因素對脂肪族氯代烴回收率的影響順序為A>B>C>D,芳香族氯代烴為A>B>D>C,催化溫度最為顯著,氫氣流速次之。脂肪族氯代烴的二次項A2、B2、D2影響顯著,C2影響不顯著;芳香族氯代烴的二次項A2、D2影響顯著,C2和B2影響不顯著。結合實際樣品處理和催化效率分析,經響應面模型分析得出最佳參數:催化溫度為260℃,氫氣流速為1.5 mL/min、催化劑用量為2 g。為進一步驗證數據的可靠性,采用上述最優條件對已知濃度的1-氯十三烷和2-氯聯苯加標混合溶液進行催化實驗,重復3次,得到催化效率分別為87.3%和92.1%,與理論值相當,所以最終采用上述條件進行實驗。

表2 Box-Behnken中心組合設計的方差分析結果Table 2 Analysis of variance results for Box-Behnken central composite design
2.5 質譜條件的選擇
將幾種氯代烴還原產物的標準溶液逐個在EI全掃描模式下以相同的電離能量進行一級質譜掃描。結果表明,C10~C17直鏈烴類物質均較易形成分子離子峰[M]+,如[C13H28]+(M=184.3)和[C16H34]+(M=226.1);也易于形成相差若干個—CH2—的 系 列 離 子 峰,如[C3H7]+(M=43.1)、[C4H9]+(M=57.3)、[C5H11]+(M=71.2)等。芳烴類物質易形成分子離子峰[M]+,如[C10H12]+(四氫萘,M=132.1)和[C12H16]+(苯基環己烷,M=160.1)等分子離子峰。分別對以上各分子離子以0~60 eV的碰撞能量進行產物離子模式掃描,結果表明,直鏈烴類物質分子離子均易形成響應最強的烷烴特征峰,[C4H9]+(M=57.3)或[C5H11]+(M=71.2)的分子離子峰,此類分子離子峰均能進一步丟失—CH2—形成分子量更低的[M-14]+烷烴或烯烴特征峰,如[C3H7]+(M=43.1)、[C3H5]+(M=41.1)和[C2H3]+(M=27.1)的分子離子;芳烴類物質易形成帶有烴基取代的苯環結構特征峰,如[C9H9]+(α-甲基苯乙烯,M=116.9)、[C8H8]+(苯乙烯,M=104.2)、[C7H7]+(甲苯,M=91.1)、[C6H5]+(苯,M=77.1)的分子離子峰。幾種氯代烴還原產物和內標物正戊基苯優化后的參數見表3,MRM總離子流色譜圖見圖4。

圖4 還原產物混合標準溶液(1.0 μg/mL)的MRM色譜圖Fig.4 MRM chromatogram of reduction products mixed standard solution(1.0 μg/mL)the peak numbers denoted were the same as those in Table 3
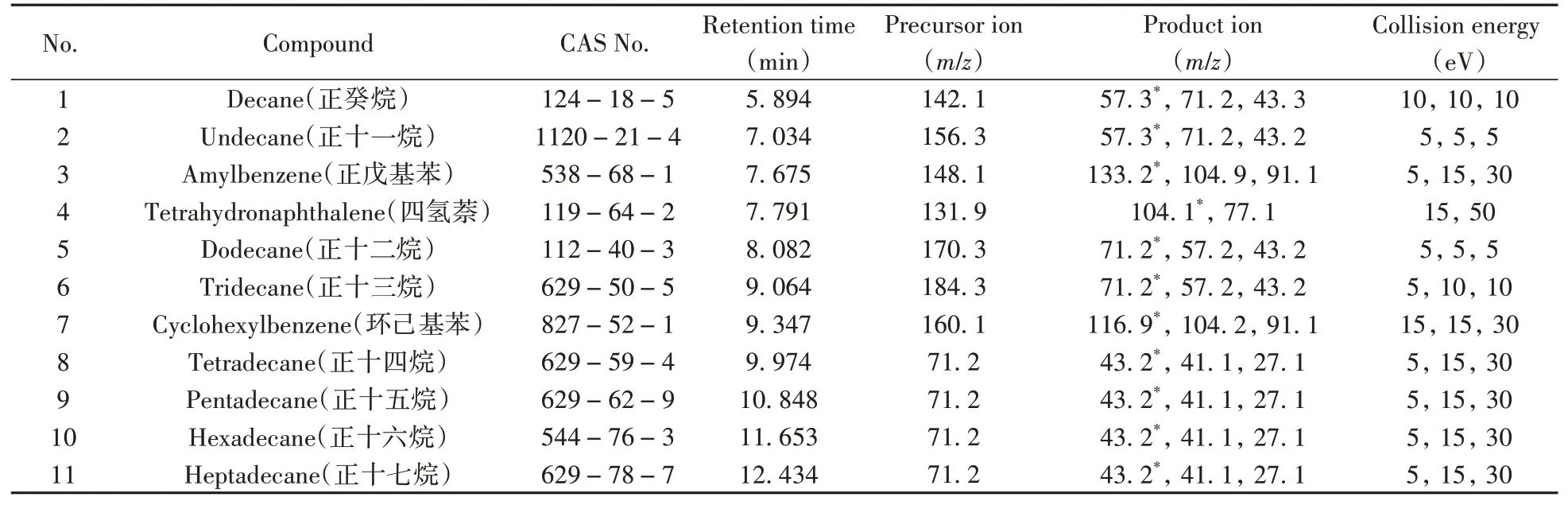
表3 還原產物及內標物優化后的定量、定性離子對和碰撞能量Table 3 Optimized parameters of quantitative ion,qualitative ion and collision energy for reduction products
2.6 線性范圍與定量下限
氯代烴目標物通過在線催化,轉化為C10~C17直鏈烷烴、四氫萘和苯基環己烷而被測定。用“1.2”的催化效率質控溶液作為測試催化劑轉化效率的質控標樣,使用還原產物標準工作溶液,以各還原產物與內標物的質量濃度比(X)和各還原產物與內標物的色譜峰面積比(Y)繪制標準工作曲線。結果表明,10種還原產物在0.02~1.00 μg/mL質量濃度范圍內線性良好,相關系數(r2)為0.995 1~0.999 3。以10倍信噪比(S/N)作為定量下限(LOQ),得到還原產物的定量下限為0.011~0.040 μg/mL,即還原產物C10~C13直鏈烷烴總和為0.098 μg/mL,C14~C17直鏈烷烴總和為0.124 μg/mL,四氫萘為0.012 μg/mL,苯基環己烷為0.011 μg/mL(見表4)。方法定量下限相當于樣品中短鏈氯化石蠟的含量為4.07 μg/kg、中鏈氯化石蠟的含量為5.33 μg/kg、多氯萘的含量為0.29 μg/kg、多氯聯苯的含量為0.26 μg/kg。
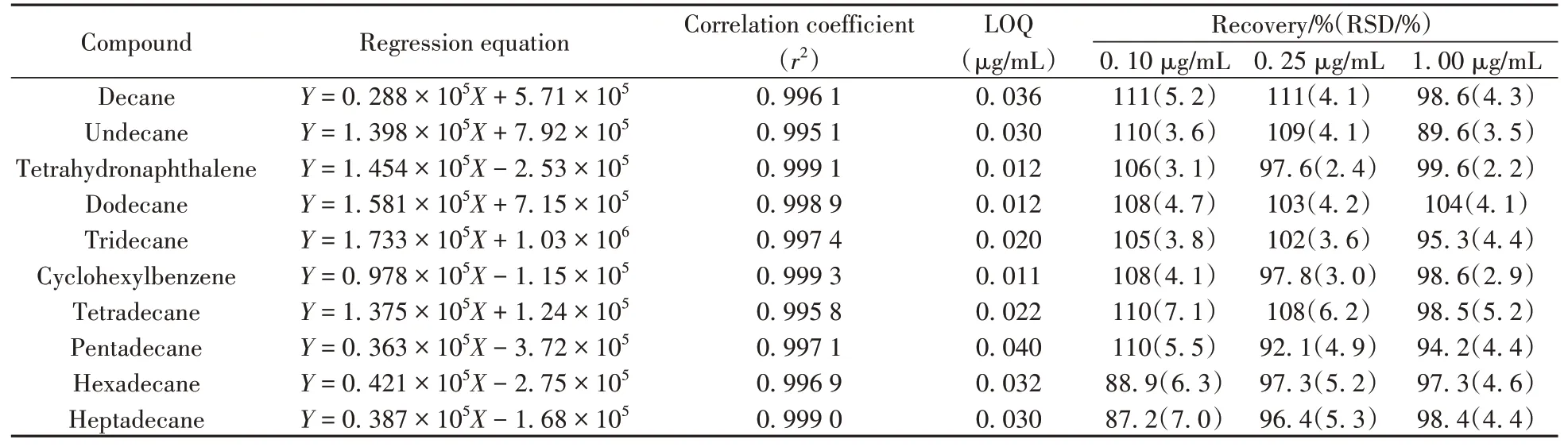
表4 還原產物的回歸方程、相關系數、定量下限、回收率和相對標準偏差Table 4 Regression equations,correlation coefficients,limits of quantitation,recoveries and relative standard deviations of the reduction products
2.7 回收率與相對標準偏差
選取不含C10~C17直鏈氯代烷烴(短鏈及中鏈氯化石蠟)、多氯萘和多氯聯苯的含油金屬原料,向其中加入適量短鏈氯化石蠟、中鏈氯化石蠟、多氯萘1014和多氯聯苯1260標準溶液,配制成目標物質量濃度均為0.10、0.25、1.00 μg/mL的基質樣液,在催化劑催化效率90%以上時測試上述3個濃度基質樣液中還原產物的濃度,每個加標水平平行測試6次,計算回收率和相對標準偏差(RSD)。由表4可知,樣品的加標回收率為87.2%~111%,RSD不大于7.1%。
2.8 實際樣品的測試
采用本方法對10批次進口含油金屬原料樣品中短鏈氯化石蠟、中鏈氯化石蠟、多氯聯苯和多氯萘的含量進行測定,每個樣品平行測試2次。結果表明,2個樣品檢出短鏈氯化石蠟、中鏈氯化石蠟和多氯聯苯。1#樣品中檢出SCCPs:152.6 μg/kg,MSCCPs:38.4 μg/kg,PCBs:0.9 μg/kg;2#樣品中檢 出SCCPs:19.3 μg/kg,MSCCPs:44.8 μg/kg,PCBs:39.4 μg/kg。某含油金屬原料樣品的MRM色譜圖見圖5。
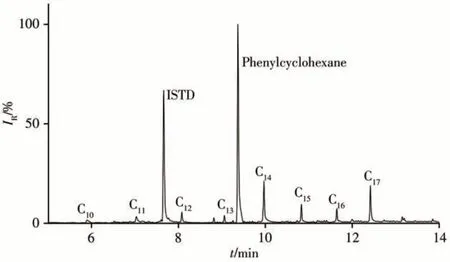
圖5 某含油金屬原料樣品的MRM色譜圖Fig.5 MRM chromatogram of an oil-bearing metal raw material
3 結論
本文運用氯代烴鈀催化技術,采用催化襯管通過在線催化/氣相色譜-串聯質譜法測定進口含油金屬原料中多氯聯苯、多氯萘、短鏈和中鏈氯化石蠟等含氯污染物的含量。目標物碳骨架通過脫氯加氫,使含氯異構體混合物轉變成C10~C17正構烷烴、四氫萘和苯基環己烷,利用氣相色譜-串聯質譜MRM模式內標法進行測定。方法減少了多氯類物質各同系物之間的干擾,從而實現了含油金屬原料中氯污染物的準確測定。該方法較為簡便、靈敏度高,能滿足進口含油再生金屬原料中含氯污染物的批量篩查和檢測需求。
猜你喜歡
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
中國塑料(2016年12期)2016-06-15 20:30:07
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
中國塑料(2016年5期)2016-04-16 05:25:36
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
中國塑料(2015年3期)2015-11-27 03:41:38
中國塑料(2015年11期)2015-10-14 01:14:14
中國塑料(2015年9期)2015-10-14 01:12:17
