扎沖十三味丸HPLC指紋圖譜研究
2023-01-12 09:52:34王會品王金梅劉丹娜
中國合理用藥探索 2022年12期
王會品,王金梅,劉丹娜
1鄭州市第三人民醫院,鄭州 450000;2河南大學藥學院,鄭州 475000
扎沖十三味丸由訶子、制草烏、木香、石菖蒲、珊瑚(制)、人工麝香、丁香、珍珠(制)、沉香、肉豆蔻、磁石(煅)、禹糧土、甘草共13味藥材制得,具有祛風通竅、舒筋活血、鎮靜安神功效,多用于半身不遂、口眼歪斜、腰腿不利、四肢麻木、筋骨疼痛、言語不清、神經麻痹、風濕及關節疼痛等[1]。方中訶子被譽為“藏藥之王”,具有抗氧化、抗病毒、抗腫瘤、抗菌和神經保護等多種藥理作用,內含活性成份沒食子酸、槲皮素和鞣花酸等[2-4]。石菖蒲的藥理活性成份為揮發油α細辛醚、β細辛醚和細辛醛等成份,多用于神昏癲癇、健忘失眠、耳鳴耳聾、脘痞不饑和噤口下痢[5-6]。甘草在成方制劑中應用廣泛,素有“十方九草”之稱,其含有的三萜類成份中以甘草酸含量最高,具有抗炎、免疫調節及抗肝損傷等生物活性,甘草含有的黃酮類成份主要有甘草素、甘草苷等,具有抑菌、抗炎、抗病毒及抗氧化等作用[7-9]。木香中含有豐富的萜類成份,主要有抗炎、抗腫瘤和抗潰瘍的作用,代表性成份有木香烴內酯和去氫木香內酯[10-11]。扎沖十三味丸收載于《中華人民共和國衛生部藥品標準》蒙藥分冊[1]中,質量標準項下未對其內在活性提出定量質控要求,相關文獻[12-14]也只是對其含有的個別成份進行考察,未見有關指紋圖譜方面報道。
中藥指紋圖譜能客觀合理、科學全面地評價中藥質量,在中藥質量分析領域應用廣泛[15-16]。本研究收集了2個不同廠家、共14批次的扎沖十三味丸樣品,建立HPLC指紋圖譜,并對指紋圖譜進行聚類分析及主成份分析,考察不同產地扎沖十三味丸的內部質量,為系統客觀、科學評價扎沖十三味丸質量提供參考依據。
1 材料
1.1 儀器
2695型高效液相色譜儀(DAD檢測器,美國Waters公司);AL204型分析天平(十萬分之一,瑞士Mettler Toledo公司);SDPP制樣粉碎機(湖南三德科技股份有限公司);KQ-600DB型超聲波清洗器(昆山超聲波儀器有限公司)。
1.2 試藥
α細辛醚對照品(成都瑞芬思生物科技有限公司,批號X-037-150921,含量≥99.0%);木香烴內酯對照品(批號:111524-201911,含量99.9%)、鞣花酸對照品(批號111959-201602,含量89.3%)、沒食子酸對照品(批號110831-201906,含量91.5%)、去氫木香內酯對照品(批號111525-201912,含量99.5%)、槲皮素對照品(批號100081-201610,含量99.1%)、甘草苷對照品(批號111610-201607,含量93.1%)、甘草酸銨對照品(批號110731-202021,含量96.2%)均購自中國食品藥品檢定研究院;乙腈(美國天地公司,色譜純);水為娃哈哈純凈水;其余試劑均為分析純。
實驗用14批扎沖十三味丸的規格均為2g/10粒。其中,S1~S8由阜新蒙藥有限責任公司生產,批號分別為:20170901、20170902、20180302、20180405、20180502、20180507、20180601、20180905;S9~S14由內蒙古大唐藥業股份有限公司生產,批號分別為:170203、170512、170605、180401、180502、181005。
2 方法與結果[13]
2.1 色譜條件
Waters Tnature C18色譜柱(250mm×4.6mm,5μm);流動相為乙腈-0.1%磷酸溶液,梯度洗脫(0~5min,5%乙腈;5~18min,5%→12%乙腈;18~25min,12%→28%乙腈;25~35min,28%→40%乙腈;35~40min,40%乙腈;40~55min,40%→58%乙腈;55~75min,58%乙腈;75~80min,58%→5%乙腈);檢測波長為254nm;流速為1.0ml/min;柱溫為36℃;進樣量為10μl。
2.2 溶液的制備
2.2.1 混合對照品溶液的制備
取本研究指認共有峰所需對照品各適量,用甲醇溶解后配制成每1ml含沒食子酸0.125mg、鞣花酸0.321mg、甘草苷0.209mg、槲皮素0.225mg、甘草酸銨0.189mg、木香烴內酯0.211mg、去氫木香內酯0.165mg、α細辛醚0.351mg的混合對照品儲備溶液。精密量取混合對照品儲備液5ml,置25ml量瓶中,加甲醇稀釋至刻度,搖勻,用0.45μm微孔濾膜濾過,取續濾液,即得混合對照品溶液。
2.2.2 供試品溶液的制備
取扎沖十三味丸,粉碎,過80目篩。取細粉約2.0g,精密稱定,置具塞錐形瓶中,精密加入50%甲醇50ml,閉塞,稱定重量,超聲處理(功率250W,頻率60kHz)1h,放冷,再稱定質量,用50%甲醇溶液補足減失重量,搖勻,用0.45μm微孔濾膜濾過,取續濾液,即得。
2.3 方法學考察
2.3.1 精密度試驗
取扎沖十三味丸供試品溶液(批號170203),按“2.1”項下所述色譜條件連續進樣6次,記錄共有峰色譜圖,以槲皮素為參比峰,計算其他24個共有峰的相對保留時間和相對峰面積。結果顯示,各共有峰相對保留時間的RSD為0.2%~0.9%,各共有峰相對峰面積的RSD為0.5%~1.2%,表明儀器精密度良好。
2.3.2 穩定性試驗
取同一批扎沖十三味丸供試品溶液(批號170203),在室溫放置0、3、6、9、12、18、24h后,按“2.1”項下所述色譜條件測定,記錄各共有峰色譜圖,以槲皮素為參比峰,計算其他24個共有峰的相對保留時間和相對峰面積。結果顯示,各共有峰相對保留時間的RSD為0.5%~1.2%,各共有峰相對峰面積的RSD為1.0%~2.0%,表明扎沖十三味丸供試品溶液在24h內穩定性良好。
2.3.3 重復性試驗
取同一批扎沖十三味丸供試品(批號170203)6份,按“2.2.2”項下方法制備供試品溶液,按“2.1”項下所述色譜條件測定,記錄各共有峰色譜圖,以槲皮素為參比峰,計算其他24個共有峰的相對保留時間和相對峰面積。結果顯示,各共有峰相對保留時間的RSD為0.4%~1.1%,各共有峰相對峰面積的RSD為0.7%~1.5%,表明本方法重復性良好。
2.4 扎沖十三味丸HPLC指紋圖譜的建立及分析
2.4.1 指紋圖譜的建立
取14批扎沖十三味丸樣品,按“2.2.2”項下方法制備供試品溶液,按“2.1”項下所述色譜條件測定,記錄各批樣品色譜圖。將14批扎沖十三味丸供試品色譜圖在同條件下積分處理后,導出AIA格式數據至“中藥色譜指紋圖譜相似度評價系統(2012版)”軟件中,設定扎沖十三味丸S1號樣品色譜圖為參考圖譜,設定時間窗寬度為0.2min,采用多點校正法對14批樣品色譜圖進行自動匹配分析,得到扎沖十三味丸對照指紋圖譜(見圖1)和14批扎沖十三味丸HPLC指紋圖譜疊加圖譜(見圖2),共確定24個共有峰。

圖1 扎沖十三味丸對照指紋圖譜

圖2 14批扎沖十三味丸樣品HPLC疊加圖譜(S1~S14)
2.4.2 相似度評價分析
設定S1號供試品色譜圖為參考圖譜,對14批扎沖十三味丸指紋圖譜進行整體相似度評價。結果,14批扎沖十三味丸供試品指紋圖譜相似度為0.960~0.993,說明不同生產廠家扎沖十三味丸指紋圖譜較為相似。相似度評價數據見表1。

表1 14批扎沖十三味丸樣品相似度評價數據
2.4.3 共有峰指認及相對峰面積計算
通過與混合對照品HPLC色譜圖(見圖3)比對,24個共有峰中共指認出8個成份,即峰5為沒食子酸、峰10為鞣花酸、峰13為甘草苷、峰15為槲皮素、峰20為甘草酸銨、峰21為木香烴內酯、峰22為去氫木香內酯、峰24為α細辛醚。其中,以峰15(槲皮素)的峰面積較大,位置適中,故設定為參照峰,計算其余23個色譜峰相對峰面積,見表2。

圖3 混合對照品HPLC色譜圖
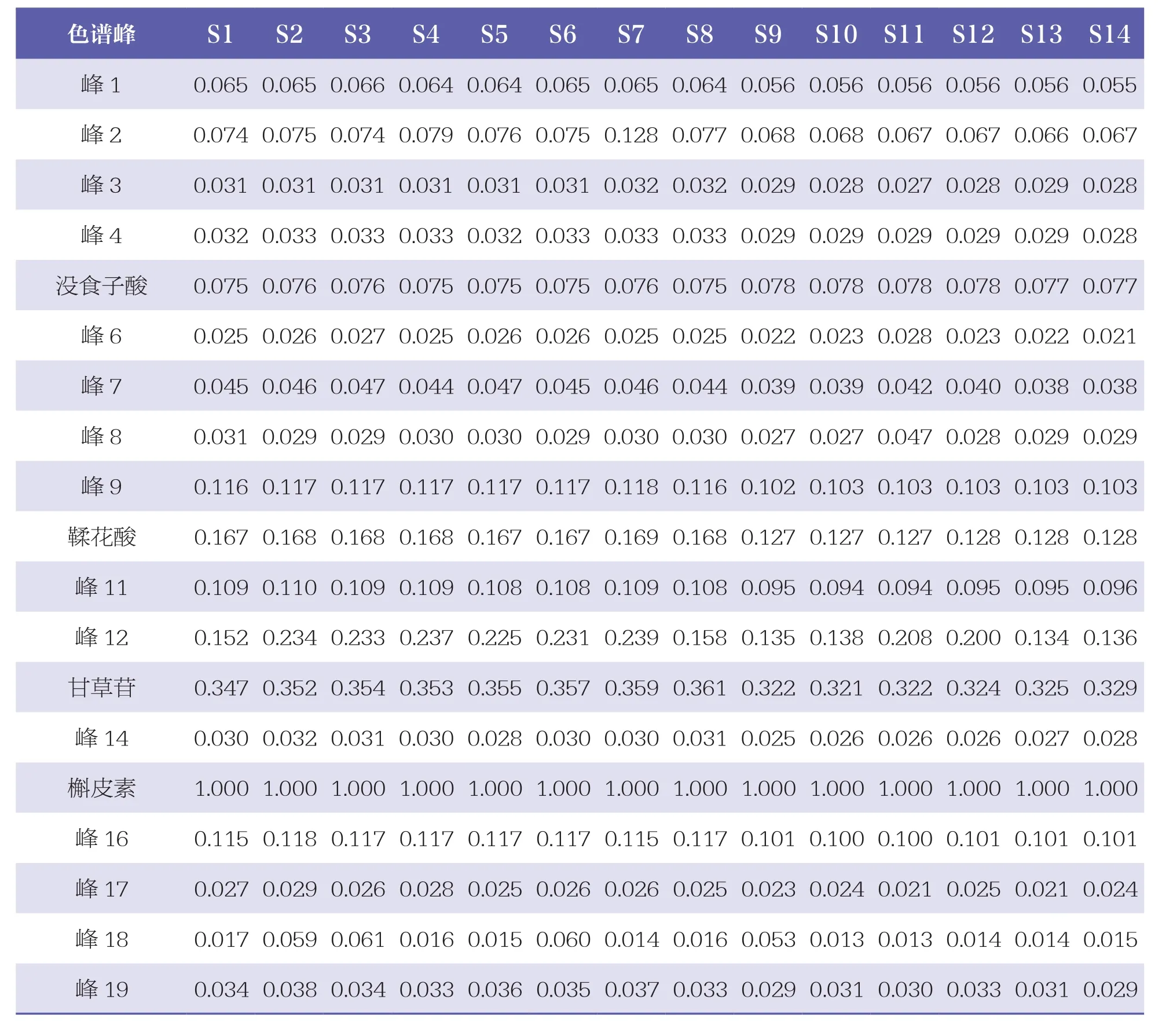
表2 14批扎沖十三味丸樣品HPLC圖譜共有峰的相對峰面積
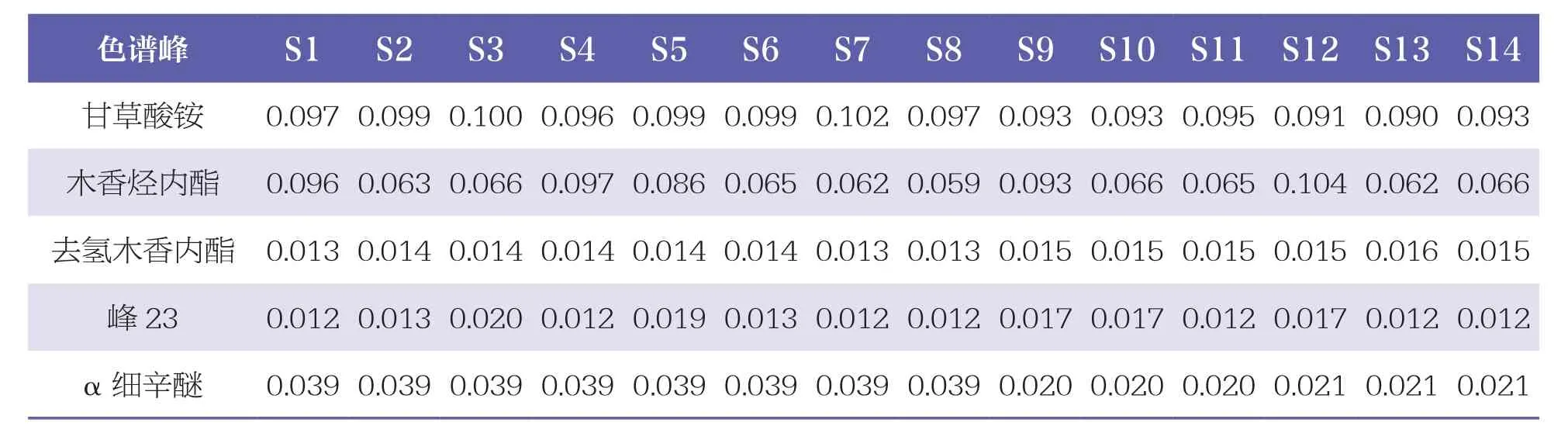
續表
2.5 聚類分析
為考察14批扎沖十三味丸樣品間差異,本研究采用SPSS 22.0軟件,以14批扎沖十三味丸指紋圖譜峰面積為變量,采用平均歐式距離法對樣本進行聚類分析,結果見圖4。由圖可知,14批扎沖十三味丸大致分為2類,Ⅰ類包括S1、S2、S3、S4、S5、S6、S7、S8,Ⅱ類包括 S9、S10、S11、S12、S13、S14。結合樣品信息可知,Ⅰ類均為阜新蒙藥有限責任公司生產,Ⅱ類為內蒙古大唐藥業股份有限公司生產。可見,不同廠家扎沖十三味丸之間相關性和相似度結果較為一致。

圖4 14批扎沖十三味丸樣品聚類分析樹狀圖
2.6 主成份分析
以扎沖十三味丸指紋圖譜中24個共有峰面積為變量,利用SPSS 22.0軟件,共提取特征值大于1的主成份3個,累計方差貢獻率為90.162%。其中,3個主成份特征值分別為19.072、1.482、1.084,其方差貢獻率分別為79.467%、6.176%、4.519%,且前3個主成份累計方差貢獻率為90.162%,提示模型預測性良好,見表3。初始因子載荷矩陣結果顯示(見表4),主成份1主要體現峰1、峰3、峰4、沒食子酸、峰6、峰7、峰9、鞣花酸、峰11、甘草苷、峰14、槲皮素、峰16、峰17、峰19、甘草酸銨及α細辛醚的信息。
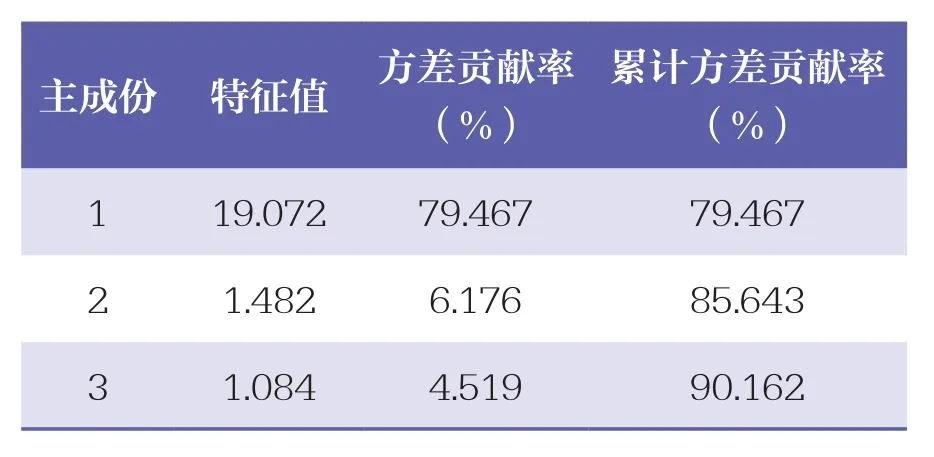
表3 3個主成份的特征值及方差貢獻率

表4 初始因子載荷矩陣

續表
用SIMCA 14.1分析軟件對14批樣品共有峰面積進行主成份分析,主成份得分圖見圖4。結果表明,14批扎沖十三味丸樣品被分為2類,與“2.5”項下聚類分析結果一致。

圖4 14批樣品主成份分析得分圖
3 討論
3.1 色譜條件的優化
結合文獻[13-14],在進行色譜條件優化過程中,以乙腈-0.1%磷酸水溶液作為流動相時,指紋圖譜峰質量較好,各目標成份均能有效分離,且信號強度較好。波長選取時,在190~400nm波段分別對沒食子酸、鞣花酸、甘草苷、槲皮素、甘草酸銨、木香烴內酯、去氫木香內酯和α細辛醚對照溶液進行全波長掃描,發現在254nm處各成份均有較強吸收,且次波長處指紋圖譜中色譜峰個數較多,所以本研究采用254nm為監測波長。
3.2 提取條件的優化
扎沖十三味丸由13味藥材制得,含有多種成份,因不同組份存在極性差異,所以樣品有效成份的提取步驟對實驗開展十分重要。為探索供試品制備方法,本實驗采用L9(34)正交試驗法,選擇甲醇濃度、溶劑用量、超聲提取時間為3個因素,以指紋圖譜的峰面積之和為指標進行考察,結果以每2g成藥粉末加50%甲醇,超聲提取1h效果最佳。
3.3 指紋圖譜分析
本研究從14批樣品的指紋圖譜中共提取到24個共有峰,通過與混合對照品色譜圖比對,指認了其中8個成份。為進一步考察扎沖十三味丸的組內和組間質量,對14批樣品進一步進行聚類分析和主成份分析,結果14批樣品聚為2類,說明不同廠家扎沖十三味丸可能因處方、工藝等因素不同而導致指紋圖譜存在差異,但同一廠家均勻性較好。在進行主成份分析時,從24個共有峰中共提取到3個主成份,累計方差貢獻率達90.162%,提示其能充分反映指紋圖譜絕大部分信息,且指認的8個成份在3個主成份中均有較大貢獻率,說明8個成份代表性較佳,可作為藥品的質控指標,用于客觀合理評價扎沖十三味丸的質量。
4 小結
中藥及其制劑均為多組份復雜體系,因此評價其質量應采用與之相適應的、能提供豐富鑒別信息的檢測方法,但現行的顯微鑒別、理化鑒別和含量測定等方法都不足以解決這一問題。建立中藥指紋圖譜能較為全面地反映中藥及其制劑中所含化學成份的種類與數量,進而對藥品質量進行整體描述和評價,這也正好符合中醫藥理論下的整體學說。扎沖十三味丸由13味藥材制成,簡單的多成份分析方式代表性欠佳,而指紋圖譜可客觀、系統地對扎沖十三味丸進行質量評價,有利于科學監管藥品的質量。本實驗首次建立了扎沖十三味丸HPLC指紋圖譜法,方法簡單、實用,方法學考察專屬性強,可有效評價扎沖十三味丸質量,為進一步完善扎沖十三味丸的標準提供了一定的實驗依據。
