堿熔-氟離子選擇電極法測定煤中氟含量
2023-01-12 06:12:20劉金祿丁仕兵張慶建
中國無機分析化學 2023年2期
關鍵詞:標準
王 旭 管 嵩 劉金祿 丁仕兵 張慶建
(1.青島海關技術中心,山東 青島 266500;2. 青島理工大學 山東 青島 266520)
氟是煤中存在的有害元素之一,主要是以無機狀態賦存于煤中,但也有少部分以有機形態存在。我國煤中氟含量一般在50~300 μg/g,少數礦區能達到3 000 μg/g。煤在燃燒過程中,氟多以HF、SiF4和CF4等形態隨煙塵排放到大氣中,受不同氣候的影響,一部分會留在空氣中,一部分則會進入土壤和水域中。人體如若攝取過多的氟元素,可能會引起一系列的病理改變和中毒癥狀。我國對煤中氟含量也做了限量規定[1],《商品煤質量管理暫行辦法》從2015年1月1號開始實行,其中規定氟≤200 μg/g,因此研究煤中氟含量的測定尤為重要[2]。
測定氟含量的方法有離子選擇電極法、分光光度法、離子色譜法和X射線熒光光譜法等方法。分光光度法[3]操作步驟復雜,對實驗化學試劑配制要求很高,容易引起測定誤差,所以靈敏度和精確度都較低;離子色譜法[4-5]應用比較廣泛,但多適用于測定氟離子濃度較低的試樣,而且采用此法很難去除煤樣中復雜基體對結果的干擾;X射線熒光光譜法[6-7]相比于其他方法操作過程簡單,但煤樣基體復雜多樣,對排除干擾要求很高;離子選擇電極法[8]具有方便快捷、精度和靈敏度高、選擇性好等特點,能消除煤樣中復雜基體成分的的干擾問題。
目前方法均需要對煤進行前處理,常用的使固體樣品中氟轉化為氟離子的前處理方法有堿熔法[9]、高溫水解法[10-13]、氧彈燃燒法等方法。氧彈燃燒法相對操作復雜,容易引入較多的測量誤差,針對灰分較高的煤樣還會出現氟元素分解不充分的現象,導致測定結果偏低[14-15]。國家標準GB/T 4633—2014《煤中氟的測定方法》,就是采用了高溫水解前處理,但其操作步驟復雜,處理過程時間較長,對操作者要求較高。因此,本文主要研究堿熔法和高溫水解法兩種前處理方法的對比情況,并對結果進行分析比較,以評估堿熔方法的可行性。
1 實驗部分
1.1 儀器及試劑
氟離子測定儀(CF-Ⅱ型,華通儀器有限公司),馬弗爐(Yamato F0410型,Japan),復合氟離子選擇電極(FL43-A0001型號,ELECTRODE),電位測定儀(Bante 931-F,般特儀器),電熱板,電磁攪拌器,瓷舟,鎳坩堝。
石英砂(分析純,國藥化學試劑公司,粒度0.5~1.0 mm),氫氧化鈉、鹽酸、硝酸、二水合檸檬酸三鈉、硝酸鉀、三乙醇胺、苯酚紅、溴甲酚綠(分析純,國藥化學試劑公司),氟化鈉(優級純,天津紅巖試劑廠),氧氣(純度99.5%以上)。
氟標準溶液(1 000 μg/mL):稱取預先在120 ℃干燥2 h的優級純氟化鈉2.210 1 g于燒杯中加水溶解,轉移至1 000 mL容量瓶中并稀釋至刻線,搖勻,移入干燥的塑料瓶中。稀釋為10 μg/mL供標準工作溶液使用。
檸檬酸鈉溶液(294 g/L):稱取294 g檸檬酸鈉加水溶解,并定容至1 L,搖勻。
三乙醇胺溶液:100 mL三乙醇胺加入64 mL鹽酸調至pH=6.5~7.0,用水稀釋至500 mL。
硝酸溶液(1+4)。
苯酚紅溶液(2 g/L):稱取0.1 g苯酚紅加入6 mL氫氧化鈉溶液(0.05 mol/L),用水稀釋至50 mL,搖勻。
煤標準物質GBW(E)110112,c(F)=72 μg/g,c(U)=10 μg/g(k=2,濟南眾標科技有限公司),煤標準物質GBW(E)110116,c(F)=122 μg/g,c(U)=10 μg/g(k=2,濟南眾標科技有限公司),煤標準物質GBW(E)110109,c(F)=158 μg/g,c(U)=11 μg/g(k=2,山東省冶金科學研究院有限公司),煤標準物質GBW(E)110108,c(F)=256 μg/g,c(U)=18 μg/g(k=2,山東省冶金科學研究院有限公司)。
1.2 樣品制備
參考標準GB/T 474—2008《煤樣的制備方法》,將煤樣經破碎、混合、縮分和干燥等步驟制備成一般分析實驗煤樣,粒度小于0.2 mm。
1.3 實驗方法
1.3.1 高溫水解法
稱取0.50 g(精確至0.000 1 g)一般分析實驗煤樣和0.5 g石英砂混勻,再用約0.5 g石英砂鋪在表面,在氧氣和水蒸氣混合氣流中于高溫燃燒水解裝置中反應,煤中氟全部轉化為揮發性氟化物溶于水中并收集于容量瓶中。加入3滴溴甲酚綠指示劑,10 mL總離子強度調節緩沖溶液,用水稀釋至刻線,搖勻靜置30 min。然后將溶液轉移至100 mL燒杯中,以氟離子選擇電極作為指示電極,用標準加入法測定溶液中氟離子濃度,從而計算得出煤中氟含量。
1.3.2 堿熔法
稱取0.50~1.00 g(精確至0.000 1 g)一般分析實驗煤樣置于鎳坩堝中,加入6 g氫氧化鈉混勻。低溫升至不同溫度并保持一段時間直至煤樣全部灰化并與堿熔融(若出現煤樣爆燃或體積膨脹嚴重則適當降低溫度延長熔融時間),后升至600 ℃保持10 min,取出稍冷后置于250 mL燒杯中,用50 mL熱水浸取,用水清洗容器至80 mL左右,加熱煮沸1 min,冷卻后將溶液和沉淀一起轉入100 mL容量瓶中,定容混勻。同時做空白實驗。
分取10.00 mL濾液置于50 mL容量瓶中,加入15 mL檸檬酸鈉溶液,1滴苯酚紅指示劑,用硝酸調至剛變黃色。加入5 mL三乙醇胺溶液,用水稀釋至刻線混勻待測。以氟離子選擇電極作為指示電極,用標準加入法測定溶液中氟離子濃度,計算煤中氟含量。
標準工作曲線:準確移取0.50、1.00、2.00、5.00、10.00 mL氟標準溶液(10 μg/mL)分別置于50 mL容量瓶中,加入10 mL空白實驗溶液、15 mL檸檬酸鈉溶液和1滴苯酚紅指示劑,用硝酸調至剛變黃色,再加入5 mL三乙醇胺溶液,用水稀釋至刻線混勻。插入電極測定每個標準溶液的電位值。以電位值為縱坐標,濃度對數為橫坐標作圖,計算出電極的斜率。
2 結果與討論
2.1 樣品量與氫氧化鈉比例
選用煤炭標準物質GBW(E)110109[c(F)=158 μg/g,c(U)=11 μg/g]作為對象,樣品量和氫氧化鈉質量比對煤中氟含量的影響如表1所示,由表1中結果可知,在一定范圍內改變稱樣量對結果影響不是很大。但是當稱樣量超過1.0 g時,測得的氟含量結果會偏低,主要是由于鎳坩堝體積有限,稱樣量較大時易產生煤樣體積膨脹過大或者從容器中濺出而不能全部與堿熔融導致部分氟的損失。當稱樣量取0.3 g時,雖然測定值在標準值不確定度范圍內,但考慮到樣品量小及分取溶液的氟含量濃度較低不在標準工作曲線范圍內,容易引起誤差,將樣品量和氫氧化鈉比例控制在0.8 g和6 g比較合理。

表1 樣品量與氫氧化鈉比例對煤中氟含量的影響
2.2 灰化溫度及時間
堿熔法中低溫灼燒溫度是測定氟含量的關鍵影響因素,圖1為不同灼燒溫度下煤標準物質GBW(E)110109[c(F)=158 μg/g,c(U)=11 μg/g]氟含量測定結果。從圖1中可以看出,溫度低于400 ℃時,氟含量均偏低,即使延長灼燒時間,坩堝內仍然有未完全灰化的煤樣,表明氟元素在此溫度范圍內不能完全釋放出來。當灼燒溫度高于400 ℃時,雖然煤樣完全灰化,但過高的溫度導致煤樣體積膨脹嚴重而與堿接觸不充分,未接觸到堿的部分在高溫下容易導致氟揮發,從而使結果偏低。綜上,低溫灼燒溫度控制在400 ℃左右時,對煤中氟含量測定最準確。

圖1 灼燒溫度對煤中氟含量測定的影響Figure 1 Effect of calcination temperature on determination of fluorine content in coal.
煤的化學組成很復雜且種類眾多,所以相同灼燒溫度下灼燒時間會有所差異,為了確保氟元素全部熔融于堿中,最終判斷依據為煤完全灰化沒有黑色粉末。
2.3 熔融溫度的選擇
低溫灰化完成后,煤中的可燃物基本上燃盡,以有機形態結合的氟化物逐步釋放出來,繼續升高熔融溫度,使煤中無機形態結合的氟化物全部釋放出來并與堿完全熔融。本實驗主要研究熔融溫度、時間對氟含量測定的影響,如表2所示,當熔融溫度為550 ℃時,測得煤標準物質的氟含量均偏低,主要是由于溫度較低時,氟不能完全釋放出來;當溫度升高到650 ℃時,過高的溫度易導致氟的揮發,氟含量也會偏低,結果表明過高或過低的熔融溫度均會引起結果偏低,而加熱時間對實驗結果影響不大。故實驗選擇控制在600 ℃熔融10 min。
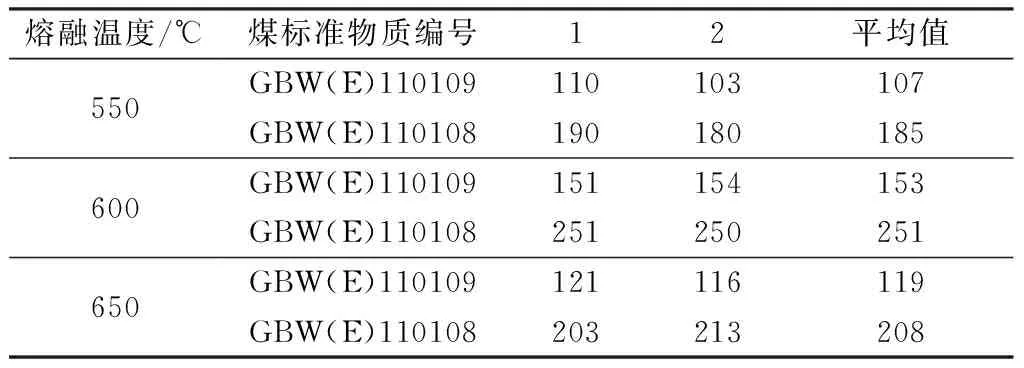
表2 熔融溫度對煤中氟含量的影響
2.4 定量分析
氟離子溶液的測定采用標準加入法:將待測溶液倒入燒杯中,插入復合氟離子選擇電極,打開攪拌器(插入深度、攪拌速度和測試溫度與測定氟電極斜率時保持一致),待電位穩定后記錄平衡電位值E1,立即加入1.00 mL氟標準溶液,待電位穩定后記錄平衡電位值E2(電位ΔE=E1-E2,選擇適當的氟標準溶液的濃度使ΔE在20~40 mV范圍內)。根據氟電極實際斜率和ΔE來計算煤中氟含量。
標準加入法的優點在于能一定程度上減小因為溶液的溫度或者所含離子成分與標準溶液不一致而引起的測定誤差,而且對于成分復雜的煤樣能消除大部分復雜基體的干擾。
2.5 標準工作曲線和檢出限
lgc與電位值E呈現良好的線性關系(c為氟濃度,μg/g),如圖2 所示,相關系數達到0.999 9,氟離子選擇電極的實際斜率為55.39(>55.0電極使用性能良好)。依據離子選擇電極法檢出限的計算方法,標準工作曲線的直線部分外延的延長線與通過空白電位且平行于濃度軸的直線相交時,其交點所對應的濃度值即為檢出限[16]。電極穩定時空白電位為449.2 mV,由此計算出檢出限為8.5 μg/g。
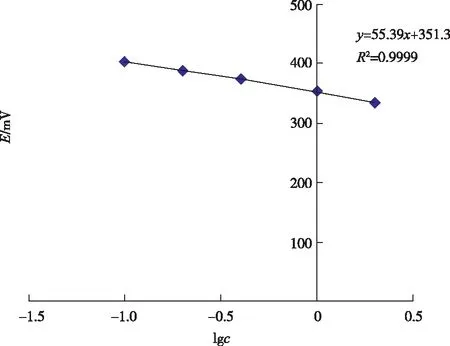
圖2 標準工作曲線Figure 2 Standard calibration curve.
2.6 方法的精密度和準確性
選用4種不同氟含量的煤標準物質進行實驗。采用高溫水解法和堿熔法兩種前處理方法,用F檢驗比較兩種方法的精密度,用t檢驗比較準確性,測定結果及檢驗結果見表3。結果表明,兩種方法間精密度沒有顯著性差異,兩種方法間測定結果沒有顯著性差異,堿熔法的測定結果和標稱值沒有顯著性差異。
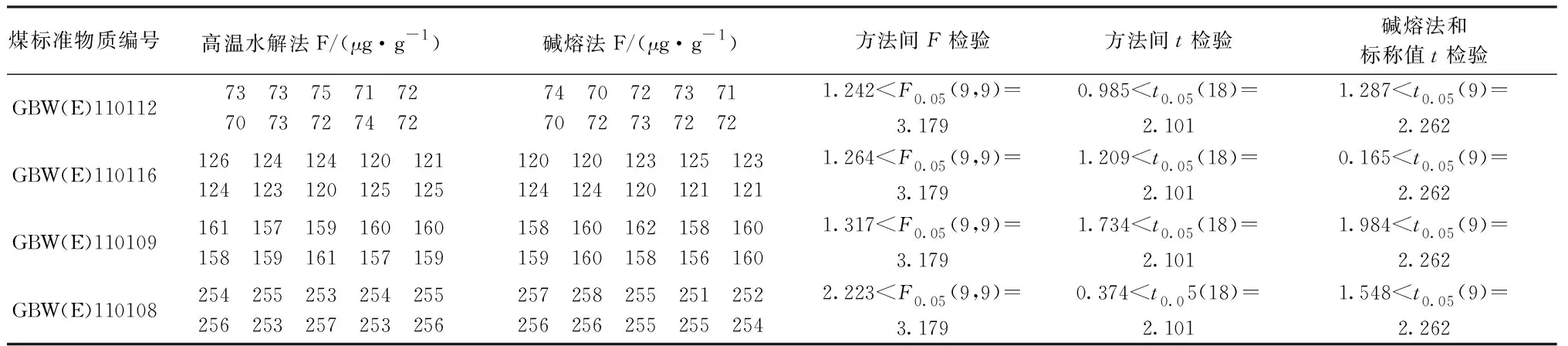
表3 氟含量測定值及統計檢驗
煤標準物質GBW(E)110116的標準值為122 μg/g,擴展不確定度10 μg/g。按照堿熔-氟離子選擇電極法,分兩批次分別進行10次重復實驗,測定結果見表4。測定結果均在不確定度范圍內,標準偏差一致,t值等于1.09 表4 堿熔方法的穩定性 1)高溫水解-氟離子選擇電極法在長期實驗中具有良好的準確度和精密度已被修訂為國家標準方法,但其冷凝液中氟的回收極易受到高溫水解過程中裝置是否達到最佳狀態的干擾,主要包括通水蒸氣時間、氧氣流量、水解溫度、水解時間等因素。在高溫水解階段,水解溫度和水解時間的控制尤為重要,為了確保燃燒溫度達到實驗要求,還需定期校準爐溫。 2)從以上結果中可以看出堿熔-氟離子選擇電極法針對不同氟含量的煤樣測定結果重復性、準確度和精密度良好。此方法主要使用到的儀器為馬弗爐,相比于高溫水解法能減少前處理過程中儀器裝置操作繁瑣帶來的誤差,而且還具有大批量處理樣品的優勢,可以解決高溫水解裝置操作繁瑣、處理費時的問題,同時樣品實現同批次處理還可以大大降低實驗環境變化對測定結果的影響。堿熔法采用低溫燃燒煤樣,能先將煤樣中的大部分有機物除去,然后堿熔的樣品用熱水即可提取完全,還可以使煤樣中的鐵、鈦、鋯、錳、稀土等干擾成分形成氫氧化物沉淀分離出去。 綜上,堿熔-氟離子選擇電極法作為煤樣氟含量測定的前處理方法,具有無需特殊裝置,操作簡便,靈敏度高,干擾少等特點,適合煤樣中氟含量的快速批量測定。
3 結論
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
當代陜西(2019年8期)2019-05-09 02:22:48
上海建材(2019年1期)2019-04-25 06:30:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
專用汽車(2016年4期)2016-03-01 04:13:43
質量與標準化(2015年9期)2015-12-31 11:41:40
中國質量與標準導報(2014年4期)2014-03-11 19:54:25
中國質量與標準導報(2014年10期)2014-02-28 22:25:47
中國質量與標準導報(2014年7期)2014-02-28 22:24:39
