基于生物信息學篩選膀胱癌預后相關的關鍵miRNAs 并驗證
2023-01-16 04:28:48鄒震海劉貝貝高五岳郭園園劉建民
醫學信息 2022年22期
鄒震海,程 琪,李 中,劉貝貝,高五岳,孫 巍,郭園園,劉建民
(蚌埠醫學院第一附屬醫院泌尿外科,安徽 蚌埠 233004)
膀胱癌(bladder cancer)是泌尿生殖系統最常見的惡性腫瘤之一。吸煙、遺傳因素以及接觸芳香胺和其它工業化學品被認為是膀胱癌發生發展的原因[1]。在所有新診斷的病例中,75%表現為非肌肉浸潤性膀胱癌(NMIBC),25%表現為肌肉浸潤性膀胱癌(MIBC)[2]。對于MIBC 患者,轉移的發生更加頻繁,預后更差,大約有50%的可能發展為轉移性疾病,其平均生存時間為14~15 個月[3],且沒有針對這些患者的治愈性治療選擇。因此,進一步研究驅動膀胱癌進展的分子機制和確定潛在的治療靶點具有重要意義。微小核糖核酸(miRNAs)是一種小的非編碼核糖核酸,長度在21~25 個核苷酸不等[4]。目前發現miRNAs 參與了哺乳動物基因組中超過50%的mRNA 的調控[5]。miRNAs 主要通過與特定目標mRNA 的3'-非翻譯區(3'-UTR)的堿基配對來調控基因轉錄,從而導致基因表達量的改變[6]。單個miRNA 能夠沉默多個基因的表達,反過來,一個基因可以被多個miRNA 調控[7]。目前,多種miRNAs 已經被證實能夠對膀胱癌細胞的生物學功能產生影響,包括細胞的增殖、侵襲、遷移和凋亡[8,9]。新一代測序(NGS)技術的快速發展和癌癥基因組圖譜(TCGA,https://portal.gdc.cancer.gov/)數據庫的建立,產生了許多大規模的癌癥基因組數據集,為利用生物信息學分析來預測相關的腫瘤生物標志物提供了基礎。本研究通過從TCGA 數據庫中獲取膀胱癌患者的miRNAs 數據及相應的預后信息,篩選出有重要預后價值的關鍵miRNAs 行下游靶基因的預測以及GO 和KEGG 通路分析,并通過實驗進行驗證,以期為膀胱癌的靶向治療提供新思路,現報道如下。
1 材料與方法
1.1 材料
1.1.1 生物信息學資料來源 從TCGA 數據庫(https://portal.gdc.cancer.gov/)中共下載418 例膀胱癌(BLCA)及19 例正常對照的miRNA 數據及相應的臨床病理特征及生存信息。臨床病理特征包括:年齡、性別、分級、分期以及T 分期。
1.1.2 材料及試劑 正常人尿路上皮細胞系SVHUC-1 以及膀胱癌細胞系T24 來源于中國科學院上海細胞庫;RPMI-1640 培養基、胎牛血清、青霉素-鏈霉素雙抗和胰酶購自美國Gibco 公司;脂質體轉染試劑Lipo2000、TRIzol、PowerUp SYBR Green Master MIX PCR 試劑盒購自美國Thermo Fish 公司;miR-NC、miR-944 mimics 和miR-1307-5p mimics 均由上海吉瑪公司設計并合成。miR-944 mimics序列:正義5'-AAAUUAUUGUACAUCGGAUGAG-3'、反義5'-CAUCCHAUGUACAAUAAUUUUU-3' ;MiR-1307-5p inhibitor 序列:正義5'-UCGACCGGACCUCGACCGGCU-3'、反義5'-CCGGUCGAGGUCCGGUCGAUU-3'。
1.2 方法
1.2.1 差異miRNAs 的獲取 利用R 軟件(v3.5.2)中edgeR 包計算差異表達的miRNAs,篩選條件:log-FoldChange=1,Padj=0.05。利用Plot 函數及Pheatmap包分別繪制火山圖及熱圖。
1.2.2 與總生存期相關的miRNAs 獲取 從TCGA 數據庫中下載BLCA 患者的臨床生存等病理信息,基于上述差異表達的miRNAs 列表,利用survival 包進行Kaplan-Meier 單因素生存分析的方法繪制不同miRNAs 高低表達的生存曲線。
1.2.3 miRNAs 與臨床病理信息之間的相關性分析將優選miRNAs 與患者5 個臨床性狀進行相關性分析(年齡、性別、分級、分期以及T 分期),利用t或Kruskal 檢驗,將miRNAs 表達量按臨床性狀分別分為兩組,得到相關性分析結果的列表。
1.2.4 預測miRNAs 靶基因及GO 和KEGG 分析 利用3 大常用的miRNA 預測靶基因的數據庫(miRDB、miRWalk、TargetScan)預測相應的靶基因,兩個及以上的數據庫都預測到的靶基因作為下一步GO 和KEGG 通路分析的基因,并繪制相應的韋恩圖。通過DAVID 數據庫(https://david.ncifcrf.gov/)對預測的靶基因進行GO 和KEGG 通路分析,以P<0.05 為差異有統計學意義;其中GO 分析包括生物過程、細胞組成和分子功能。
1.2.5 細胞培養及轉染 正常人尿路上皮細胞系SVHUC-1,以及膀胱癌細胞系T24 細胞置于含10%胎牛血清的RPMI-1640 培養基中培養。培養箱條件37 ℃、5% CO2。當細胞處于對數生長期時,0.25%胰酶消化傳代,每2 d 換液或傳代1 次。在其生長狀態良好時,將T24 細胞接種于6 孔板中,生長至60%融合度,按照Lipofectamine 2000 試劑說明書,將miR-NC、miR-944 mimics 和miR-1307-5p mimics轉染入細胞。
1.2.6 CCK8 細胞增殖實驗 將轉染后的T24 細胞接種于96 孔板中,分別培養24、48、72 h 后,每孔加入10 μl CCK8 溶液,避光,37 ℃條件下孵育1 h,酶標儀檢測細胞在450 nm 處的吸光度值(OD450 nm 值)。
1.2.7 Transwell 小室法 細胞侵襲實驗中,上室制備Matrigel 膠后,將細胞加入無血清培養基中,吸取100 μl 加入上室。下室加入含20%胎牛血清的RPMI-1640 培養基。37 ℃恒溫培養箱中培養24 h后,輕輕去除上室中的細胞和Matrigel 膠,用4%多聚甲醛固定30 min 后結晶紫染色,顯微鏡下拍照并分析。細胞遷移實驗中,Transwell 上室不加入Matrigel 膠,其余步驟同細胞侵襲實驗。
1.2.8 流式細胞術 收集轉染后的細胞,取5~10 萬重懸的細胞,1000 g 離心5 min,棄上清。加入PBS 重懸后,1000 g 離心5 min,以195 μl Annexin V-FITC結合液重懸細胞。加入5 μl Annexin V-FITC、10 μl碘化丙啶染色液,混勻后室溫孵育10~20 min,流式上機檢測。
1.2.9 總RNA 提取及qRT-PCR 根據說明書,使用Trizol 試劑從細胞中分離總RNA,將RNA 逆轉錄成互補脫氧核糖核酸(cDNA),PowerUp SYBR Green Master MIX PCR 試劑盒用于測定miRNA(U6 作為內參基因)的表達水平。2ΔΔCt 法用于計算目的基因的相對表達量。使用Primer 5.0 設計引物,見表1。
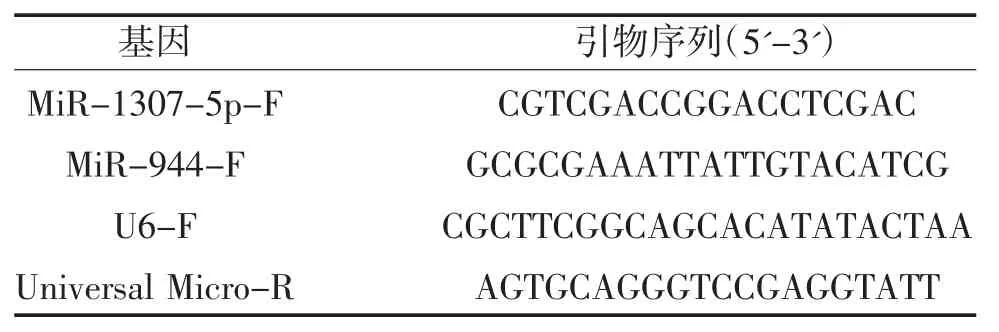
表1 引物序列
1.3 統計學方法 基因表達數據通過log2轉換標準化,使用R 軟件3.6.2 和Perl 語言包構建圖,另使用SPSS 22.0 統計學軟件和GraphPad Prism9 進行實驗數據分析處理,計量資料以(±s)表示,組間比較采用t檢驗。以P<0.05 為差異有統計學意義。
2 結果
2.1 膀胱癌差異miRNAs 的篩選結果 通過對從TCGA 數據庫中共下載的418 例膀胱癌及19 例正常對照的miRNA 數據進行分析,共篩選出293 個差異表達的miRNAs,其中高表達224 個,低表達69個,繪制火山圖見圖1。
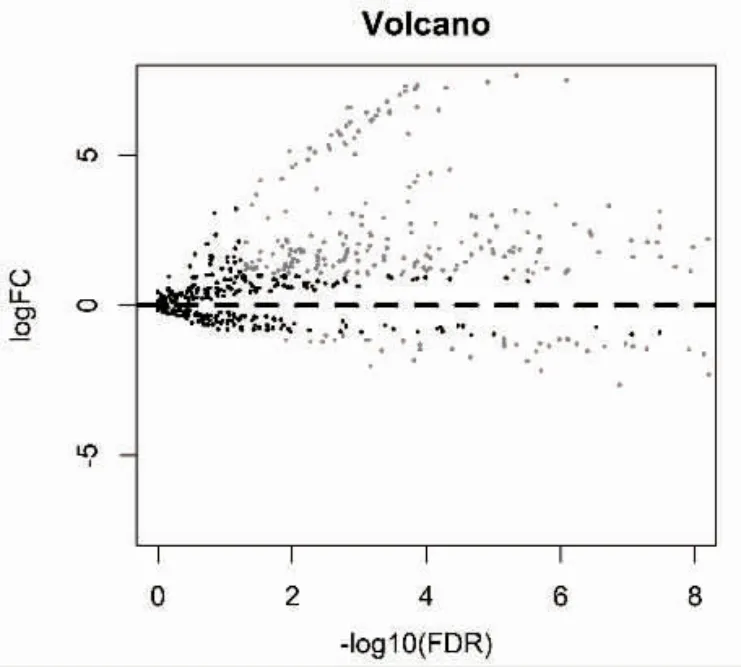
圖1 差異表達miRNAs 的火山圖
2.2 差異表達的miRNAs Kaplan-Meier 單因素生存分析 基于上述293 個差異表達的miRNAs,利用survival 包進行Kaplan-Meier 單因素生存分析的方法繪制不同miRNAs 高低表達的生存曲線,共得到65 個OS 生存曲線中P值<0.05 的miRNAs,根據P值大小,選擇表現出重要預后價值的前9 個miRNA(圖2)。Kaplan-Meier 生存曲線分析顯示,miR-502b-3p、miR-1307-5p、miR-1910-5p、miR-944 的高表達和miR-483-5p、miR-483-3p、miR-615-3p、miR-217-5p、miR-708-3p 的低表達將有利于膀胱癌的預后。
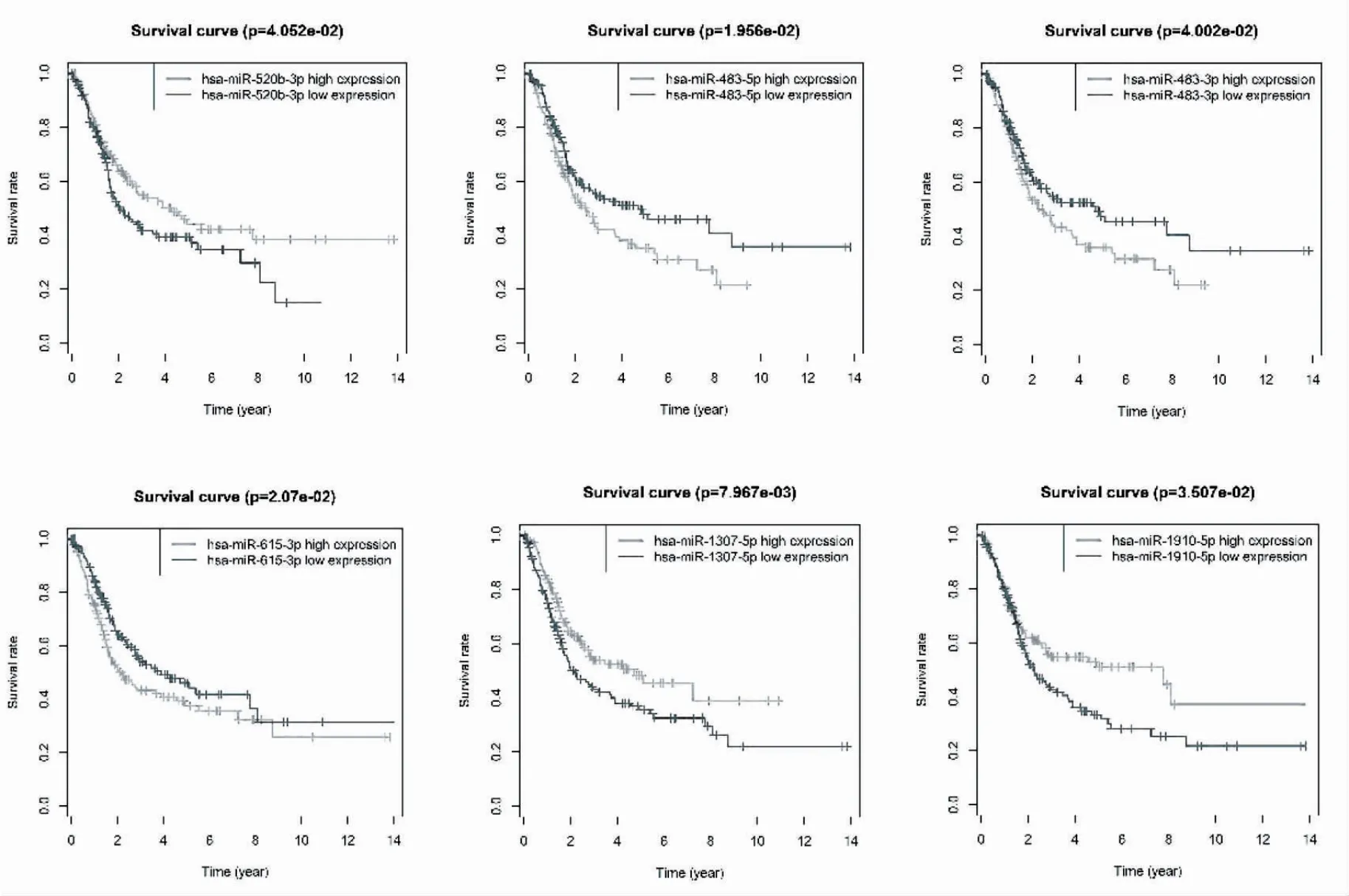
圖2 9 種miRNAs 的OS 生存曲線圖

圖2 9 種miRNAs 的OS 生存曲線圖(續)
2.3 miRNAs 與臨床病理信息之間的相關性分析 將優選的9 個miRNAs 與患者5 個臨床性狀進行相關性分析(年齡、性別、分級、分期以及T 分期),利用Kruskal 檢驗,將9 個miRNAs 表達量按臨床性狀分別分為兩組,并得到相關性分析結果的列表(表2)。根據P值,共有2 種miRNA 與膀胱癌的臨床病理信息相關,分別是miR-1307-5p 和miR-944,其中miR-1307-5p 與患者年齡、分級、分期以及T 分期相關,miR-944 與患者分級、分期以及T 分期相關(P<0.05)。
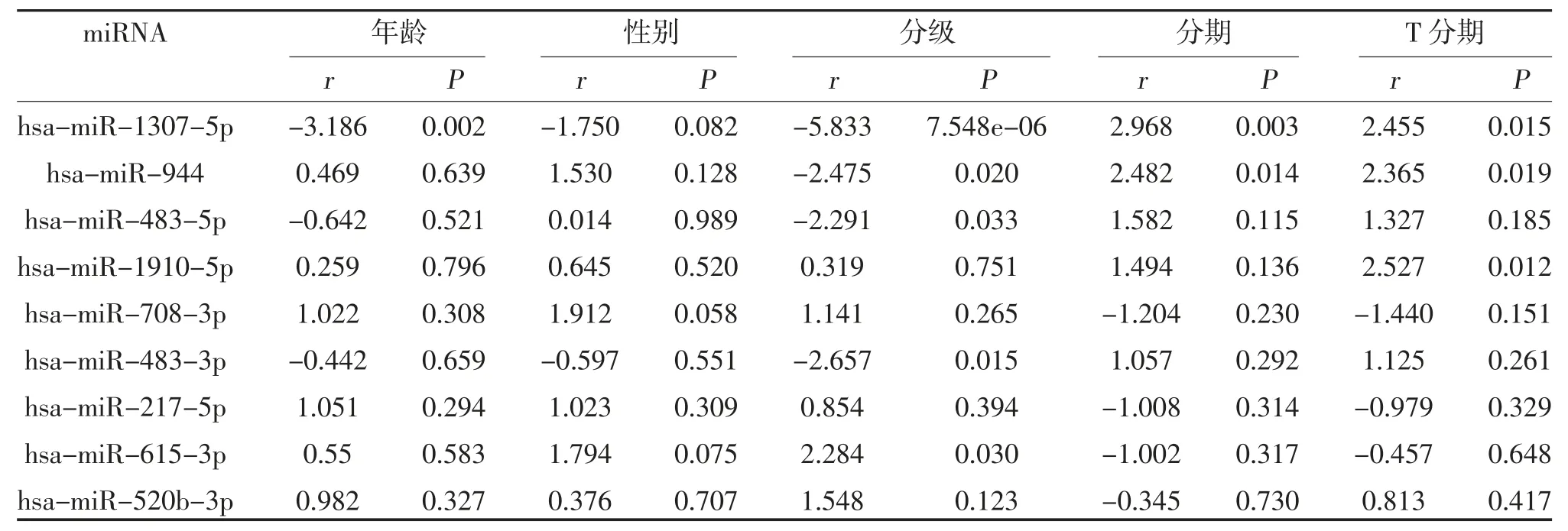
表2 膀胱癌中優選的9 個miRNAs 的相關性分析結果
2.4 差異表達的關鍵miRNAs 靶基因的預測分析利用3 大常用miRNA 預測靶基因的數據庫(miRDB、miRWalk、TargetScan)預測相應的靶基因,兩個及以上的數據庫都預測到的靶基因作為下一步GO 和KEGG 通路分析的基因,去除9 個重復的基因,總共留下1669 個靶基因用于進一步的GO 和KEGG 通路分析,并繪制相應的韋恩圖,見圖3。
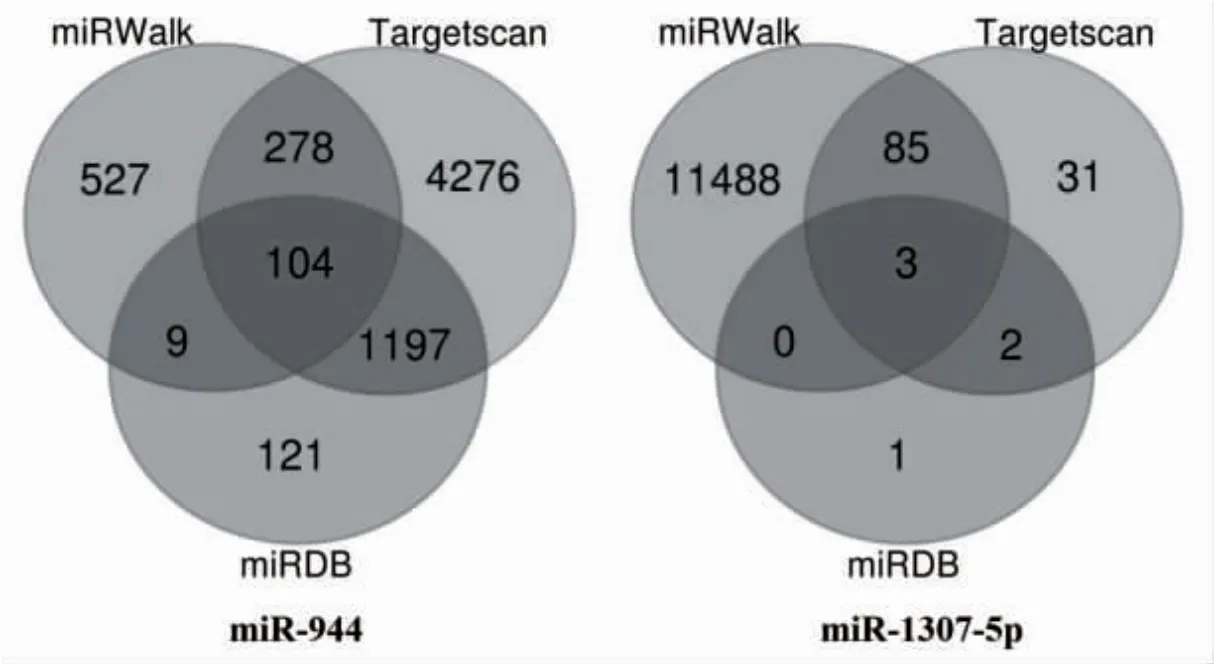
圖3 3 大miRNA 靶基因數據庫預測的靶基因
2.5 預測靶基因的GO 和KEGG 通路分析 通過DAVID 數據庫對預測的靶基因進行GO 和KEGG通路分析,GO 分析顯示,預測的靶基因在生物過程中主要富集于神經系統發育、多細胞生物發育、解剖結構發育、單細胞-多細胞生物過程和發育進程,細胞組成中主要富集于細胞內、細胞內成分、細胞器、細胞內細胞器和細胞質,分子功能中主要富集于離子結合、陽離子結合、金屬離子結合、轉移酶活性和蛋白質絲氨酸,見表3;KEGG 通路分析顯示,預測的靶基因主要富集于長壽通路、mTOR 信號通路和EGFR 酪氨酸激酶抑制劑耐藥性等通路中,見表4、圖4。

圖4 miRNA 靶基因的KEGG 信號通路分析
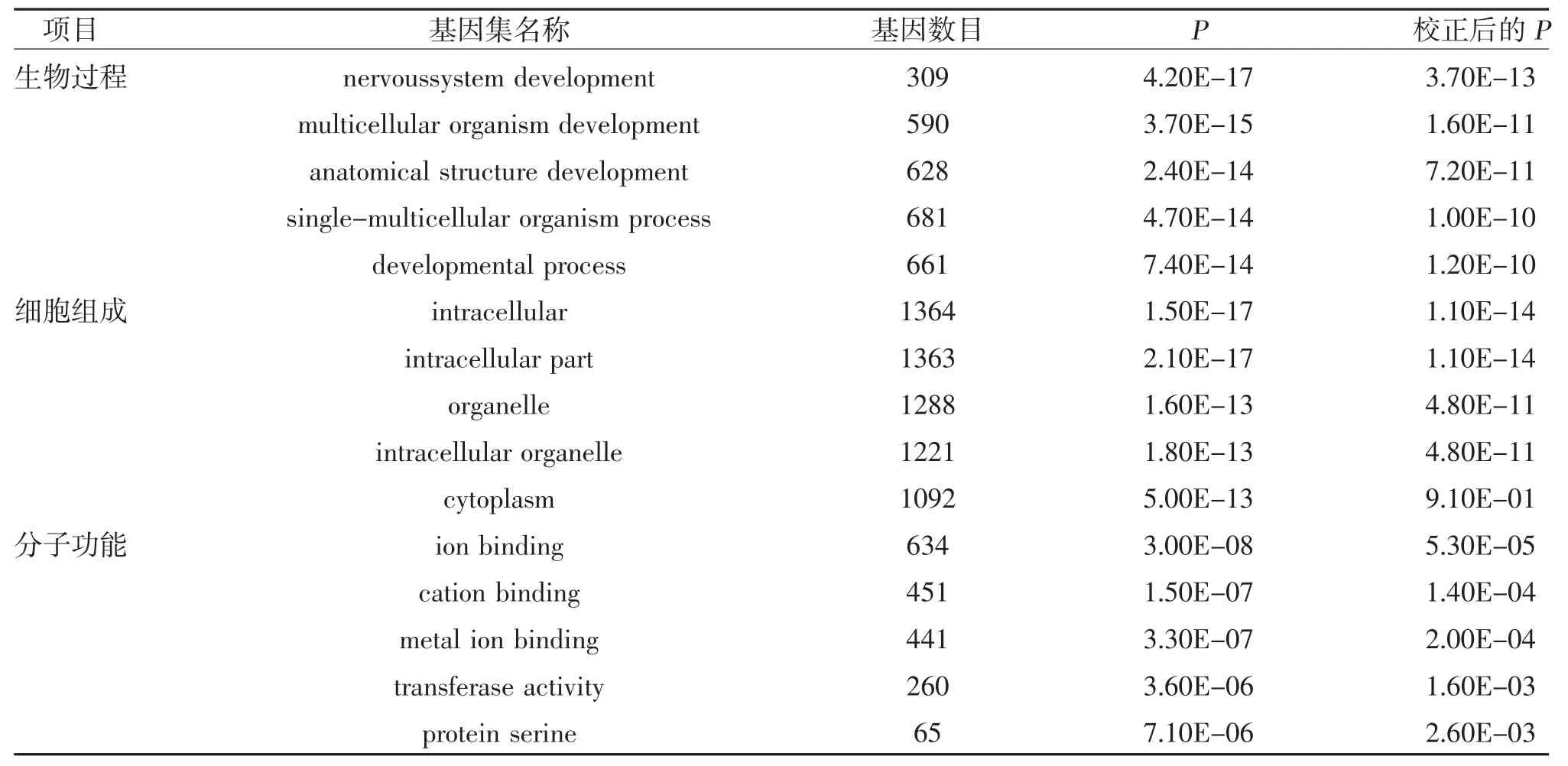
表3 排名前5 位的miRNA 靶基因的GO 功能富集分析
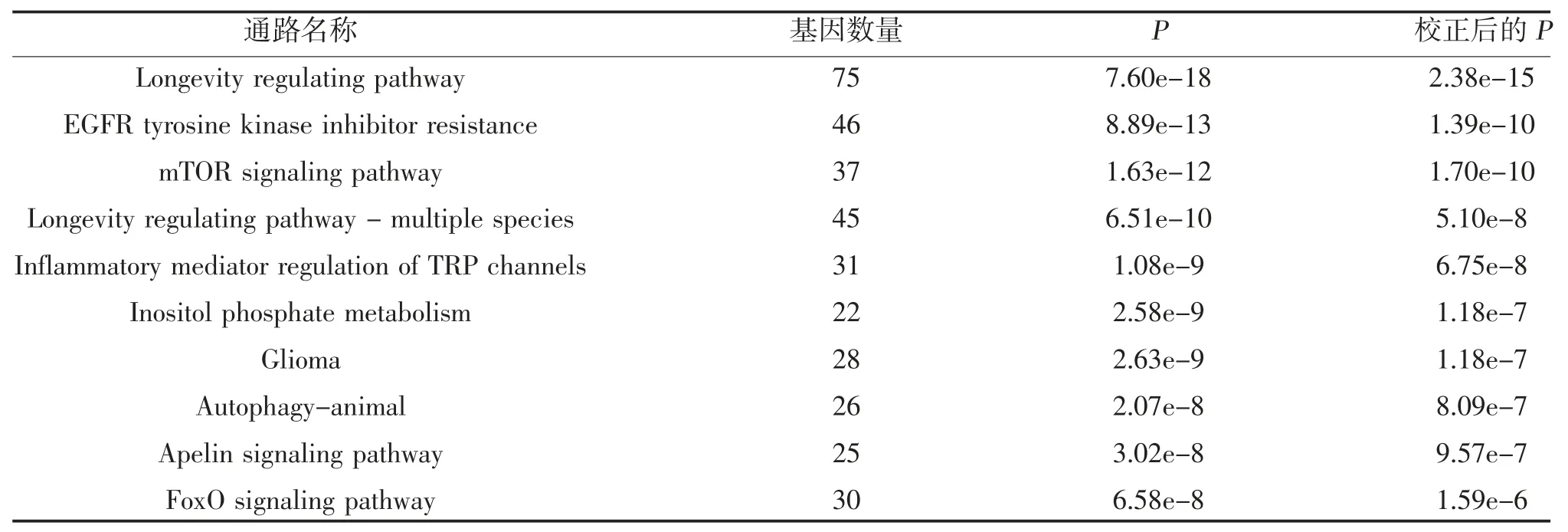
表4 排名前10 位的miRNA 靶基因的KEGG 信號通路富集分析
2.6 miR-944 和miR-1307-5p 在膀胱癌細胞中的表達水平 與正常尿路上皮細胞SV-HUC-1 相比,膀胱癌T24 細胞中miR-944 的表達水平下降,差異有統計學意義(P<0.05),而miR-1307-5p 的表達水平無變化(P>0.05),見圖5。
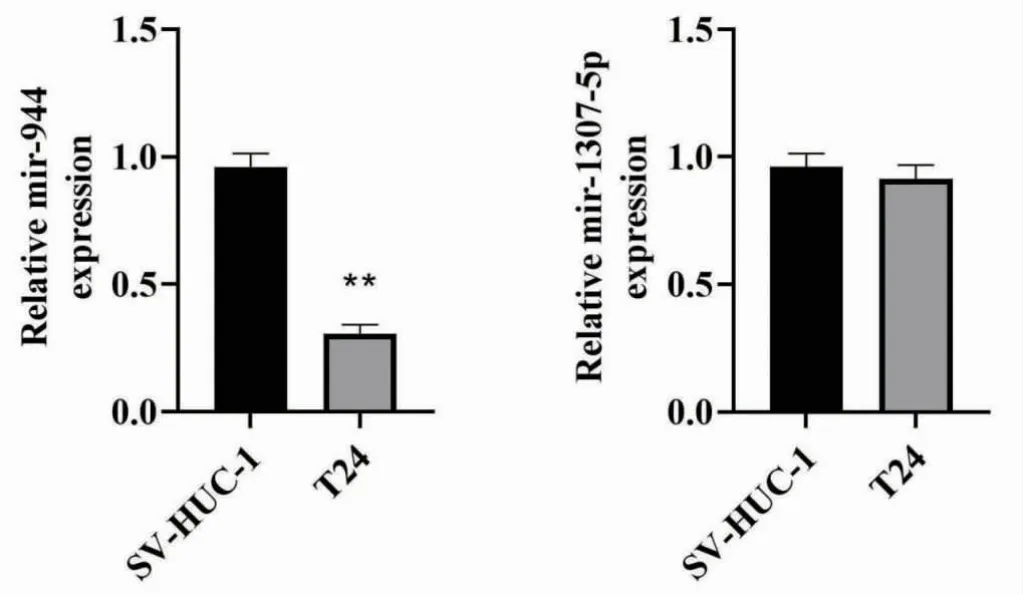
圖5 miR-944 和miR-1307-5p 在膀胱癌細胞中的表達水平
2.7 過表達miRNA 對膀胱癌細胞增殖、遷移、侵襲及凋亡的影響 CCK8 增殖實驗顯示,與mimics-NC 組相比,miR-944 mimics 組的T24 細胞的增殖能力減弱,差異有統計學意義(P<0.05),見圖6A;Transwell遷移和侵襲實驗顯示,miR-944 mimics 組的T24 細胞的遷移和侵襲能力均減弱,差異有統計學意義(P<0.05),見圖6B;流式細胞術分析顯示,miR-944 mimics 組的T24 細胞凋亡增加,差異有統計學意義(P<0.05),但過表達miR-1307-5p 后膀胱癌細胞的增殖、遷移、侵襲和凋亡無明顯變化,見圖6C。

圖6 miR-1307-5p 和miR-944 對膀胱癌細胞增殖、遷移、侵襲及凋亡的影響

圖6 miR-1307-5p 和miR-944 對膀胱癌細胞增殖、遷移、侵襲及凋亡的影響(續)
3 討論
膀胱癌是全球第9 大常見的惡性腫瘤,每年大約有356 000 例新增病例和145 000 例死亡病例,具有復發傾向,需要在診斷后終生監測[10,11]。miRNAs是一類短的非編碼RNA,能夠與目標mRNA 上的互補序列結合,通過抑制蛋白質的翻譯或增加mRNA的降解來誘導基因沉默[12]。目前miRNAs 的異常表達已被證明與多種人類疾病有關[13]。Yin Z 等[14]研究發現,miR-96-5p 能夠負性調控抑癌基因FOXO3,促進乳腺癌細胞的增殖和侵襲,從而發揮促癌作用。Chen Y 等[15]研究證明,miR-148a 通過靶向Wnt1 抑制肺癌細胞的遷移和侵襲,這可能為肺癌轉移的分子機制提供新的思路。然而,還有許多miRNA 在膀胱癌中發揮著抑癌或促癌的作用,其功能和機制仍需進一步研究。因此,本研究對TCGA 數據庫中下載的418 例膀胱癌患者及19 例正常對照的miRNAs數據進行生物信息學分析,利用R 軟件中edgeR 包獲取差異表達的miRNAs,共篩選出293 個差異表達的miRNAs,其中高表達224 個,低表達69 個。再利用survival 包進行Kaplan-Meier 單因素生存分析,選擇表現出重要預后價值的前9 個miRNAs,最后通過臨床病理信息的相關性分析得到2 個密切相關的miRNAs,分別是miR-1307-5p 和miR-944。
研究表明[16],miR-1307-5p 和miR-944 與腫瘤的發生發展均密切相關。miR-1307-5p 是通過修飾miR-1307 前體的5' 端粒獲得的一種成熟體miRNA,其可通過靶向ING5 的表達來提高卵巢癌細胞的化療耐藥性。Du X 等[17]通過一系列體內外實驗表明,miR-1307-5p 也能夠促進肺腺癌增殖,該作用機制是通過靶向TRAF3 上調MAPK/NF-κB 通路實現的。miR-944 最早通過miRNAs 克隆法在人宮頸細胞中鑒定獲得[18],該miRNA 位于腫瘤蛋白p63(TP63)基因的內含子中,定位于人3q28 染色體上[19]。miR-944 的異常表達在人類惡性腫瘤中發揮抑癌或致癌的作用。研究表明[20],miR-944 在結直腸癌患者中的表達水平顯著降低,并且通過靶向下游調節因子MACC1 抑制結直腸癌細胞的遷移和侵襲,這表明miR-944 可能在結直腸癌中發揮抗轉移作用。但miR-944 在宮頸癌中又顯著過表達,并通過靶向HECT 結構域連接酶W2(HECW2)和S100P 結合蛋白(S100PBP)促進細胞增殖、遷移和侵襲[21]。然而,在膀胱癌中,這兩種miRNAs 的具體作用有待進一步的驗證。
為了更深入地了解這2 種miRNAs 的分子機制,3 大常用miRNA 預測靶基因的數據庫(miRDB、miRWalk、TargetScan)被用于預測相應的靶基因并利用DAVID 數據庫對預測的靶基因進行了GO 富集和KEGG 通路分析。GO 分析顯示,預測的靶基因在生物過程中主要富集于神經系統發育、多細胞生物發育、解剖結構發育、單細胞-多細胞生物過程和發育進程,細胞組成中主要富集于細胞內、細胞內成分、細胞器、細胞內細胞器和細胞質,分子功能中主要富集于離子結合、陽離子結合、金屬離子結合、轉移酶活性和蛋白質絲氨酸;KEGG 通路分析顯示,預測的靶基因主要富集于長壽通路、mTOR 信號通路和EGFR 酪氨酸激酶抑制劑耐藥性等通路中。以上結果說明這兩種miRNA 可能通過影響膀胱癌細胞內的細胞質和細胞器的功能以及干擾金屬陽離子等其他物質的結合發揮相應的作用。最后,相關分子生物學技術和細胞實驗進一步驗證了生物信息學分析的結果,qRT-PCR 分析顯示miR-944 在膀胱癌細胞中的表達水平下調,而miR-1307-5p 的表達水平無明顯差異。同時,細胞功能學實驗也證實了miR-944 對膀胱癌細胞生物行為的抑制作用,這與上述qRT-PCR 的結果一致。
總之,本研究利用來自TCGA 數據庫中膀胱癌患者的miRNAs 數據和預后信息,分析了膀胱癌組織和正常膀胱上皮組織之間的miRNAs 表達以及各項臨床病理數據,獲得了2 種異常表達的miRNAs。通過進一步的分析,預測了miRNAs 的下游靶基因和靶基因富集的信號通路,最后進行相關實驗驗證后發現miR-944 有潛力成為預測膀胱癌診斷、治療和預后的一種新的生物標志物。
猜你喜歡
音樂探索(2022年2期)2022-05-30 21:01:37
民用飛機設計與研究(2020年4期)2021-01-21 09:15:02
小天使·一年級語數英綜合(2019年8期)2019-08-27 02:23:00
電子制作(2018年18期)2018-11-14 01:48:24
小學科學(學生版)(2018年7期)2018-08-13 09:33:04
財經(2017年2期)2017-03-10 14:35:35
山東工業技術(2016年15期)2016-12-01 05:31:22
財經(2016年15期)2016-06-03 07:38:02
財經(2016年3期)2016-03-07 07:44:46
財經(2016年6期)2016-02-24 07:41:51
