陸地棉RIL群體產量與纖維品質性狀QTL定位
2023-01-16 02:41:14黃莎張正圣王文文劉大軍
西南大學學報(自然科學版) 2023年1期
黃莎, 張正圣, 王文文, 劉大軍
西南大學 農學與生物科技學院,重慶 400715
棉花作為全球重要的經濟農作物,是最多的天然纖維來源,被廣泛應用于織造行業.栽培棉花多為異源四倍體,由兩個異源二倍體棉屬經A基因組與D基因組雜交后染色體加倍形成[1].陸地棉(Gossypiumhirsutum)作為最重要的異源四倍體棉種占領了超過全球95%的棉花市場.產量和纖維品質是棉花研究中最受關注的部分[2],但棉花產量與纖維品質呈負相關,難以同時改良[3],是培育優異棉種的巨大挑戰.棉花產量及纖維品質作為數量性狀,受多個基因控制.探究棉花產量與纖維品質的遺傳決定因素,利用分子標記檢測來為目的基因定位提供依據,對棉花品質改良具有重要意義.由于陸地棉種內雜交多態性低,因此構建高密度的陸地棉種內遺傳圖譜是其分子標記輔助選擇育種的關鍵.
構建高密度遺傳圖譜需要尋找眾多的分子標記.簡單序列重復(Simple sequence repeat,SSR)標記分布多且廣,實驗重復性好且結果可靠,可以在等位基因間顯示多個差異[4-5].簡化基因組測序(Specific locus amplified fragment sequencing,SLAF-seq)是在高通量測序的背景下發展起來的[6-7].SLAF-seq的3個顯著優勢是:深度測序保證了基因分型準確; 測序成本低; 正式測序前利用預測系統提升標記效率.SLAF-seq近年來已被廣泛用于單核苷酸多樣性(Single nucleotide polymorphism,SNP)檢測[8-11].
數量性狀位點(Quantitative trait loci,QTL)是在調控生物體數量性狀中起重要作用的基因片段在染色體上的位置,數量性狀不僅受多個QTL的影響,也與環境互作相關,部分數量性狀存在一個主效基因對該性狀的控制起主導作用,農作物的產量性狀一般為數量性狀.對棉花而言,產量性狀、纖維品質和抗逆性等都是數量性狀.CottonQTLdb搜集了大量來自全球棉花纖維品質、產量性狀、抗病性、耐鹽等各類性狀的QTL,在release 2.3版本中囊括了截至2018年1月來自156個刊物的4 892個QTL[12-13]; Ijaz等[14]總結了2017-2019年多個研究者定位的、與纖維品質相關的穩定QTL.
本研究運用SLAF-seq SNP技術結合SSR分子標記構建高密度遺傳圖譜,幫助增加遺傳圖譜的準確性,縮小了QTL長度區間,使QTL定位更加準確高效,可有效推進后續精細定位群體選擇和候選基因篩選,為陸地棉產量和纖維品質的分子育種提供參考.
1 材料與方法
1.1 材料
雜交母本為渝棉1號,由西南大學棉花研究室自主培育而成,產量高、纖維品質好.父本為超早3號,由中國農業科學院棉花研究所提供,具有早熟、無腺體、株型緊湊等特點.重組近交系(RIL)F2:6群體(包含184系)及兩親本材料分別于2019年夏種植于重慶歇馬棉花種植基地,2019年冬種植于海南三亞,2020年夏種植于新疆庫爾勒和新疆奎屯.
1.2 DNA提取與SLAF-seq 測序
提取2019年夏重慶歇馬棉花種植基地的棉花植株嫩葉DNA,提取過程參照Zhang等[15]改良的CTAB(Cetyltrimethylammonium Bromide)法.將提取的184株棉花全套DNA送至北京百邁客生物科技有限公司進行SLAF-seq測序.參考棉花基因組來自http://ibi.zju.edu.cn/cotton/,利用北京百邁客生物科技有限公司自主研發的基因組酶切軟件對DNA進行酶切預測,選擇HaeIII+Hpy166II作為最適內切酶組合,經酶切得到的片段范圍長度在414~464 bp之間,預測有254 089個SLAF(Specific-Locus Amplified Fragment)標簽,經日本晴酶切顯示,Control數據雙端比對效率為88.74%,酶切效率為84.23%,構建的SLAF庫合格.
1.3 SSR分子標記
本研究篩選引物包括CCRI,SWU,JESPR,Gh,NAU,BNL,CIR,CGR,HAU,DOW,DPL[16].引物CCRI的設計參照亞洲棉基因組[17],引物SWU的設計參照雷蒙德氏棉基因組[18],其他來源于棉花引物數據庫http://www.cottonmarker.org.PCR反應體系:1.5 μL 10x Easy Taq PCR Buffer(Mg2+)、0.2 μL 2 mmol/L dNTP、0.3 μL 25 mmol/L Mg2+、0.2 μL 10 mmol/L前引物(pF)、0.2 μL 10 mmol/L后引物、再用ddH2O定容至10 μL.PCR反應流程:預變性94 ℃ 5 min、35個循環(變性94 ℃ 30 sec、退火53 ℃ 30 sec、延伸72 ℃ 30 sec)、延伸72 ℃ 7 min、保溫 16 ℃ 30 sec.采用聚丙烯酰胺凝膠電泳法篩選多態性SSR引物,并利用篩選得到的具多態性引物對群體進行基因型檢測.
1.4 遺傳圖譜構建與QTL定位
將測序結果進行篩選,并將基因型缺失大于80%的標記去除,利用JoinMap 4[19]軟件對經SSR分子標記和簡化基因組測序得到的SNP標記進行聯合分析,作圖映射函數為Kosambi函數[20],去除共分離后的位點用于構建遺傳圖譜,參照Li等[21]測序得到的陸地棉基因組長度計算各染色體的基因組覆蓋率.
利用MapQTL?6.0[22-24]對群體進行QTL定位,導入所有環境位點基因型數據和表型數據,選擇區間作圖法運算得到每個環境QTL的遺傳距離,設置似然比對數LOD≥2.5的位點作為確定的QTL鄰近標記,LOD值減1的區間作為置信區間,將多個環境檢測到的同一性狀、同一位置的QTL記為同一個QTL,QTL的命名方式為“q性狀-染色體-序號”.利用MapChart將得到的QTL定位在遺傳圖譜上.QTL簇的命名方式為“qClu-染色體-序號”.
1.5 數據統計分析
鈴質量(BW)、衣分(LP)、籽指(SI)、衣指(LI)按常規方法測定; 纖維上半部平均長度(FL)、纖維整齊度指數(FU)、纖維斷裂比強度(FS)、纖維馬克隆值(FM)、纖維伸長率(FE)送至中國農業科學院棉花研究所進行纖維檢測.各性狀間的相關性系數圖由R語言繪制:https//www.R-project.org/.
2 結果與分析
2.1 產量和纖維品質表型統計分析
4個環境下9個性狀均表現出不同程度的超親分離現象.頻率分布直方圖顯示各性狀均近似正態分布,且不同環境間性狀表現存在差異,結合4個環境的9個表型性狀進行方差分析得到F值(表1),表明這些性狀連續分布且易受環境影響,符合數量性狀遺傳特點.重組近交系F2:6群體在2019年夏重慶(CQ)、F2:7群體在2019年冬海南(HN)、F2:8群體在2020年夏新疆庫爾勒(KL)和新疆奎屯(KT)4個環境中的表型數據如表2所示.相關性分析表明,纖維整齊度、纖維斷裂比強度、纖維伸長率都與纖維長度呈顯著正相關,纖維斷裂比強度、纖維伸長率都與纖維整齊度呈顯著正相關,纖維伸長率與纖維斷裂比強度.籽指和衣分兩兩呈顯著正相關,纖維長度、纖維斷裂比強度都與馬克隆值呈顯著負相關(圖1).

表1 4個環境間產量與纖維品質的方差分析
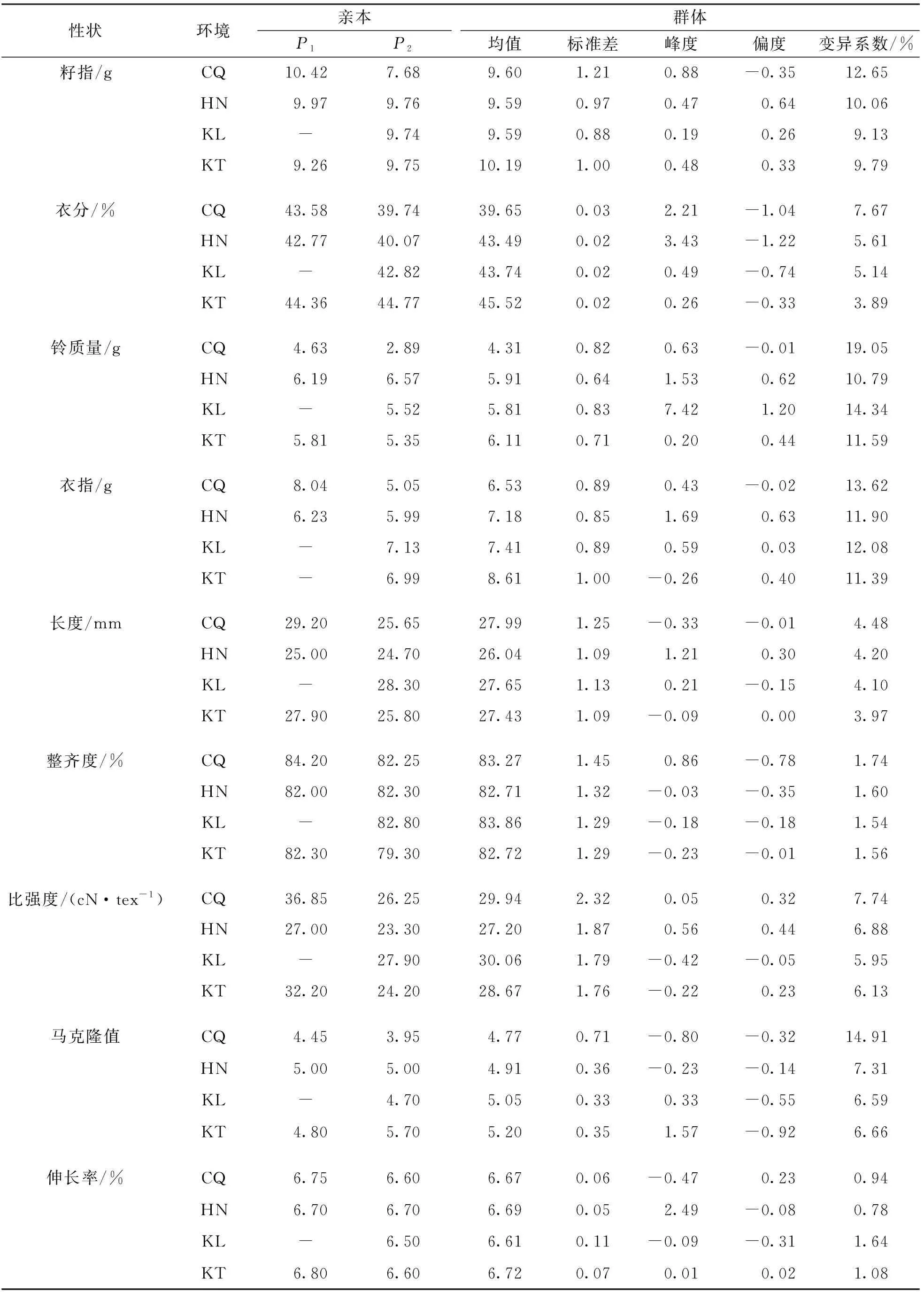
表2 親本及RIL群體的產量和纖維品質性狀表現
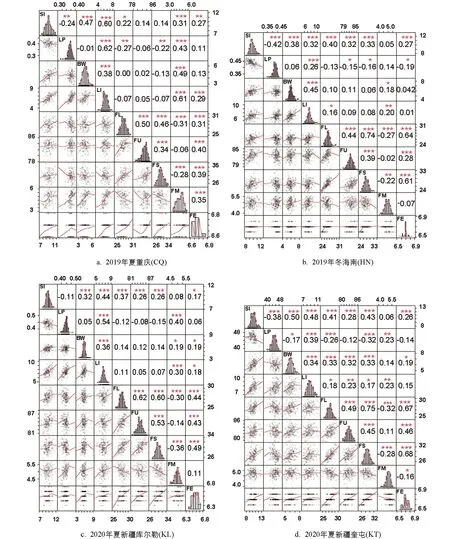
圖1中數據由對RIL群體4個環境9個性狀統計得出,經R語言分析,可以表示出每個單株的具體性狀統計值及性狀間的相關性.對角線為該環境各性狀的頻率分布直方圖及擬合曲線; 對角線左側顯示帶有擬合曲線的二元散點圖,每個點表示一個單株,其橫縱方向對應的數值為對應性狀的統計值; 對角線右側顯示兩個性狀間的相關性系數及顯著性水平,***表示p<0.1%; **表示p<1%; *表示p<5%水平差異具有統計學意義.圖1 RIL群體產量和纖維品質性狀表現及性狀相關性
2.2 遺傳圖譜構建
利用3 578對SSR引物對親本進行多態性篩選得到有多態性的引物145對,多態性比率為4.05%.SLAF-seq測序報告顯示共有49 528個SNP標記(圖2).過濾掉測序缺失率大于80%和顛倒錯位的標記.經SLAF-seq SNP結果和SSR分子標記結果共同分析,最終得到分布于26條染色體的8 020個標記,包括60個SSR分子標記和7 960個SNP標記.經過分離過濾后,構建了包含2 945個位點,總遺傳長度為4 650.71 cM,位點間平均遺傳距離為1.58 cM的遺傳圖譜,物理長度為2 202.95 Mb.對比TM-1參考基因組的2 240.95 Mb,本文構建的遺傳圖譜基因組覆蓋率為98.30%(表3).
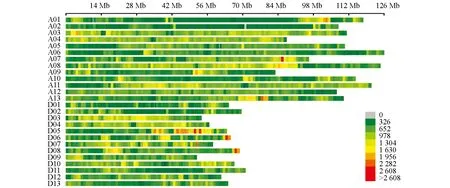
圖2 SNP密度分布圖
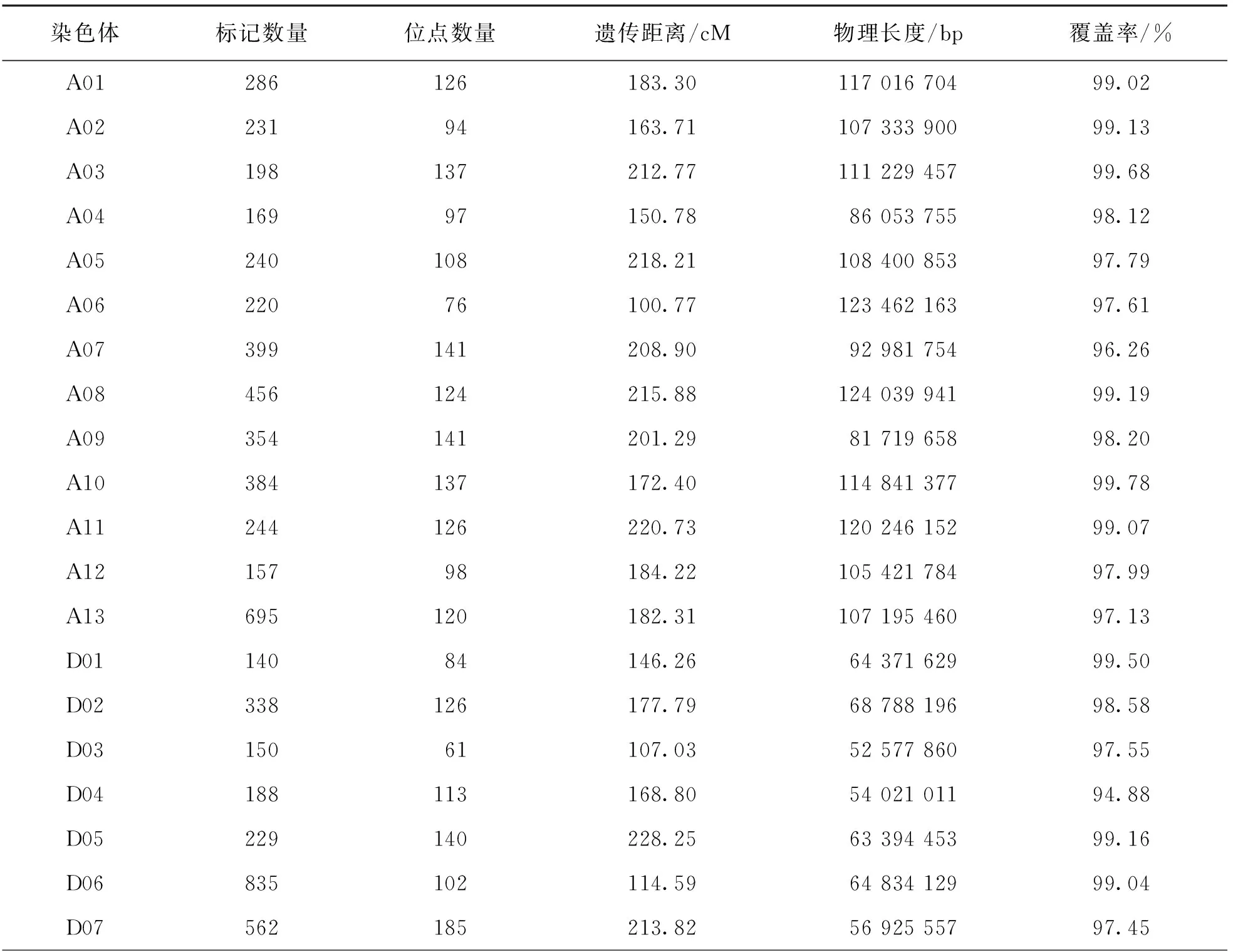
表3 遺傳圖譜標記在染色體上的分布情況
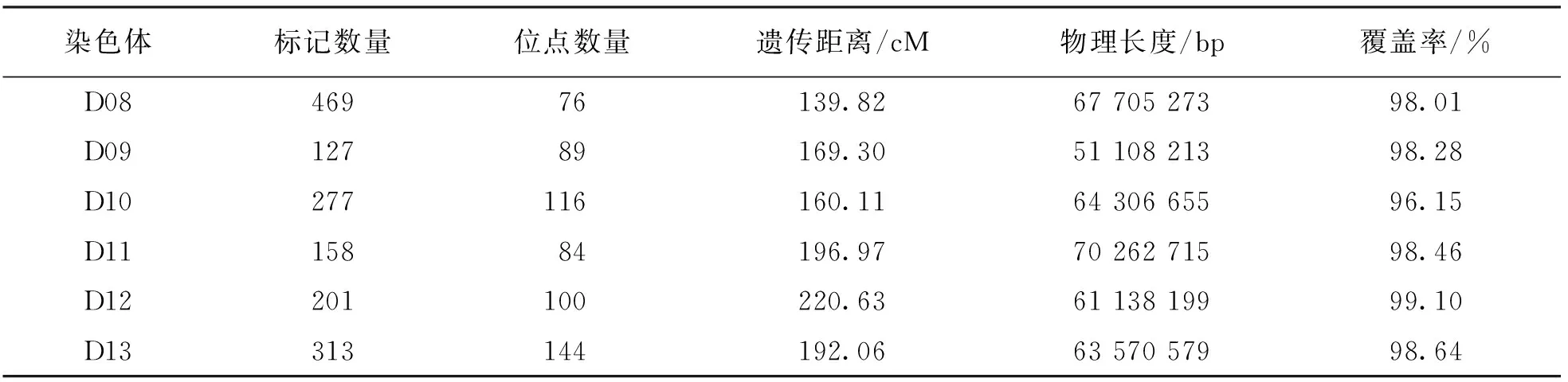
續表3
2.3 QTL定位
結合高密度遺傳圖譜與4個環境的產量及纖維品質性狀統計數據共同分析,本研究共定位到分布于26條染色體上的76個QTL,包括35個產量性狀QTL,41個纖維品質性狀QTL,LOD值分布在2.50~7.76之間,解釋表型變異值為6.4%~23.4%.當解釋表型變異值大于10%時,視為主效QTL.加性效應值顯示,QTL增效基因來源于渝棉1號的有41個,來源于超早3號的有35個.有10個QTL在兩個及以上環境中被檢測到(表4).纖維長度性狀的QTL qFL-D11-1在4個環境中均被檢測到.在4條染色體上共檢測到5個QTL簇,包含17個QTL(圖3).
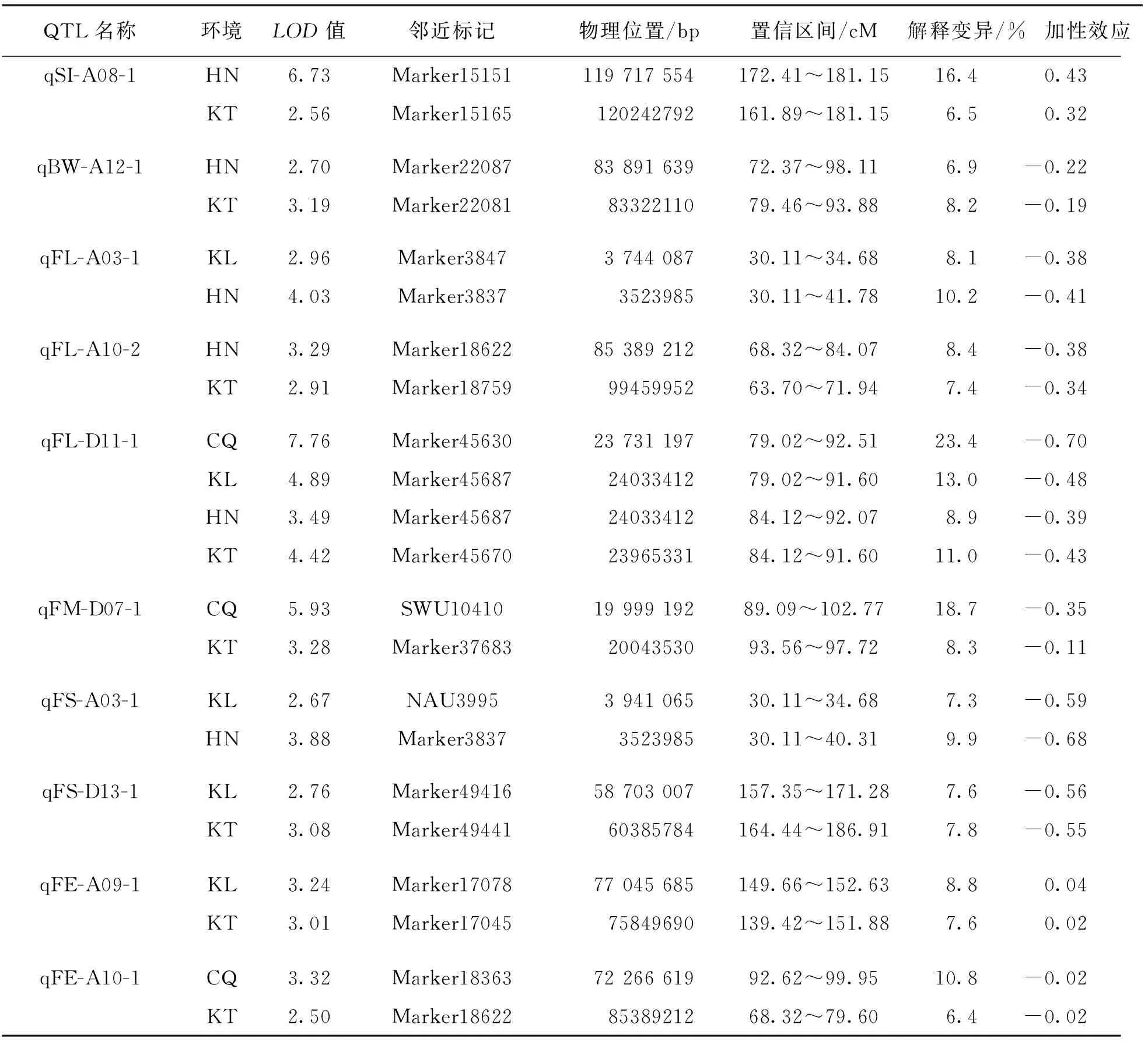
表4 在兩個及以上環境中存在的QTL
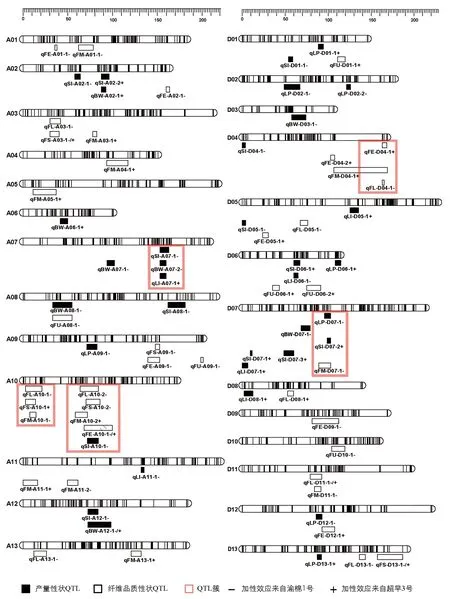
圖3 遺傳圖譜及產量性狀和纖維品質性狀QTL定位圖
3 討論
3.1 遺傳圖譜構建
分子遺傳圖譜的構建已經在植物的分子生物學研究中有了廣泛的應用[25-27].分子標記是構建遺傳圖譜的前提,在RFLP(Restriction fragment length polymorphism),RAPD(Random Amplified Polymorphic DNA),AFLP(Amplified Fragment Length Polymorphism)等眾多的傳統標記方式中,本研究選擇了廣泛應用的SSR分子標記技術,該技術準確度高,可重復利用[28],但工作量大且耗時長.SNP高多態性配合SLAF-seq技術高效且價格相對便宜的優勢,成為分子標記的新選擇.在測序結果分析時,篩除了SNP標記中基因型缺失大于80%或者位置明顯錯誤的標記,導致部分區域分子標記存在較大間隙.本研究結合了SSR分子標記和SLAF-seq SNP技術,且兩者相互補充,提高了遺傳圖譜構建的準確性.
3.2 QTL定位
高密度遺傳圖譜是QTL定位的有力保障,多個環境及多個性狀的測量使QTL定位更加準確.在眾多QTL定位研究中,定位到的QTL數量差異很大,是因為遺傳群體類型不同,親本遺傳背景不同,分子標記類型不同,遺傳圖譜密度不同,多數表型性狀受環境影響大等各類差異所致[29-31].研究定位到的QTL增效基因來源不同且QTL加性效應來源也不一致,表明在子代表型統計時出現了超親分離現象.本研究結合高密度遺傳圖譜與多環境性狀定位到的76個產量和纖維品質性狀QTL,可以為棉花品質精細定位群體的構建提供依據.在41個纖維品質QTL中,關于纖維長度的QTL qFL-D11-1在4個環境中均被檢測到,且其LOD值為所有QTL中的最大值7.76,可為后續棉花纖維品質的圖位克隆提供重要參考.
3.3 QTL簇
QTL成簇是在棉花、水稻、玉米等許多農作物的QTL定位中都存在的現象[32-34].關于棉花QTL成簇現象,置信區間內存在“一因多效”現象的解釋被廣泛認可,Ulloa等[35]指出棉花的重要農藝性狀QTL在染色體上表現出高度重組和基因富集的現象; Rong等[36]提出QTL簇的出現還可能是纖維相關QTL代表了一組具有新功能的小基因家族,棉花纖維發育過程的變異涉及復雜的網絡或相互作用的基因.本研究過程中發現當遺傳圖譜標記間隔較大,定位到的QTL置信區間較大時,QTL之間易出現區間交叉形成QTL簇,因此高密度遺傳圖譜的構建尤為重要.本研究在A07染色體上的QTL簇qClu-A07-1包含了籽指、衣分、鈴質量3個產量性狀的QTL,且加性效應均來自渝棉1號,表明該位點可實現對陸地棉的3個性狀同時改良.
4 結論
本研究構建了一張高密度遺傳圖譜,包含2 945個上圖位點,總遺傳長度為4 650.71 cM,圖譜物理總長為2 202.95 Mb,覆蓋基因組總長的98.30%.定位到76個QTL,包括產量性狀QTL 35個,纖維品質性狀QTL 41個,LOD值分布在2.50~7.76之間,解釋表型變異率為6.4%~23.4%.本研究可為后續精細群體構建及候選基因篩選奠定基礎,為早熟棉的產量與纖維品質相關育種提供參考.
