先天自愈性朗格漢斯組織細胞增生癥一例
2023-01-21 05:01:04周桂芝
中國麻風皮膚病雜志 2023年1期
王 娜 吳 梅 周桂芝 楊 青
山東第一醫科大學附屬皮膚病醫院(山東省皮膚病醫院),山東省皮膚病性病防治研究所,濟南,250022
先天性自愈性朗格漢斯組織細胞增生癥(congenital self healing Langerhans cell histiocytosis,CSHLCH)臨床少見,臨床表現多樣,可表現為丘疹、結節、結痂、水皰,較少出現出血性大皰,有時出現潰瘍,甚至軟疣樣丘疹。
臨床資料患兒,女,1個月。頭面部紅色丘疹、結痂,逐漸增多1個月。患兒出生時有輕度的羊水污染,出生后頭皮、額頭、軀干有藍紫色和深紅色斑,之后頭面部出現散在出現黃豆至豌豆大紅褐色至黃褐色丘疹、結節。生長發育史:足月剖宮產,生長發育正常,母乳喂養,父母體健,無家族遺傳病史。
體格檢查: T 36. 5℃,P 125次/min,R 30次/min,體重4.5 kg。全身淺表淋巴結未觸及腫大,心肺無異常,肝脾未觸及腫大。皮膚科情況: 頭皮、額頭、軀干、四肢散在黃豆至豌豆大紅褐色至黃褐色丘疹、丘皰疹、結痂,部分皮疹少量鱗屑,不易刮除(圖1) 。
實驗室及輔助檢查: 血常規、尿常規、肝腎功能無異常。胸部X線及腹部B超肝脾胰腎未見異常。皮膚共聚焦顯微鏡示:皮損區域角化過度、角化不全,囊腔形成,大量的炎細胞移入,真皮淺層血管輕度擴張。皮損組織病理示: 表皮細胞間水腫,單一核細胞移入,基底細胞液化變性,真皮淺中層較多單一核細胞浸潤,細胞核大、胞漿豐富(圖2a)。DIF:表皮細胞間及基底膜帶IgG、C3、IgM、IgA陰性。免疫組化:CD1a、S100、CD68弱陽性,Langerin陽性(圖2b)。診斷: 先天性自愈性朗格漢斯組織細胞增生癥。未予治療。
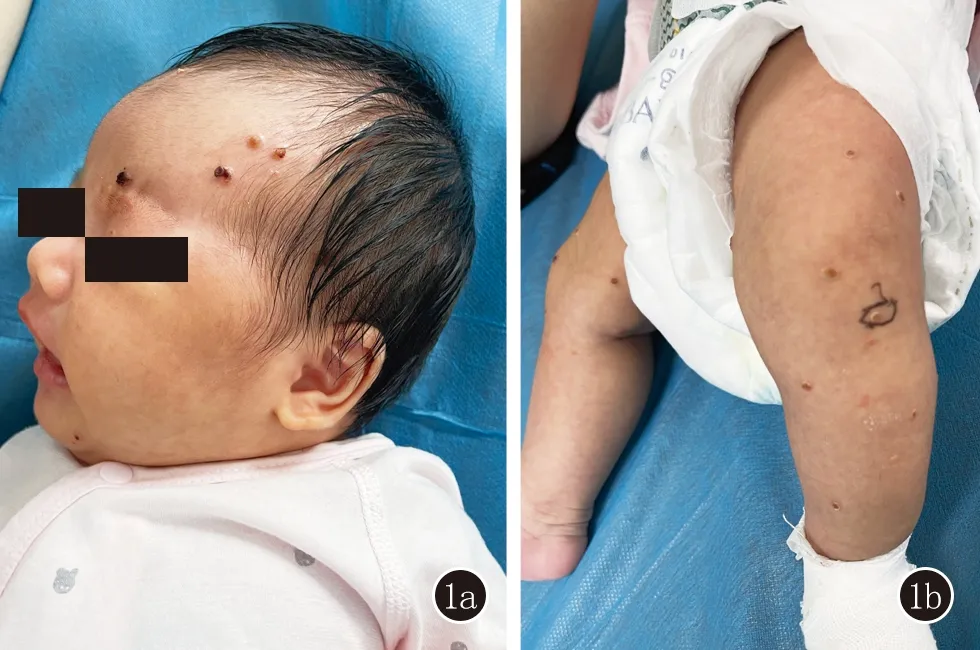
圖1 頭皮、額頭(1a),四肢(1b)散在黃豆至豌豆大紅褐色至黃褐色丘疹、丘皰疹,局部結痂
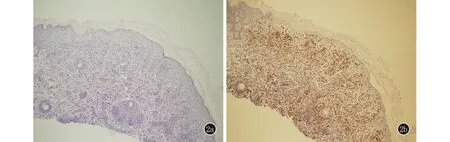
圖2 2a:表皮細胞間水腫,單一核細胞移入,基底細胞液化變性,真皮淺中層較多單一核細胞浸潤,細胞核大、胞漿豐富(HE,×100);2b:Langerin陽性(免疫組化,×100)
隨訪: 3個月內原皮疹自行消退,無新皮疹出現(圖3)。患者行BRAF V600E 野生型,全外顯子測序陰性,CNV測序發現1處罕見基因組拷貝數變異:Seq[GRCh37]dup(16)(q22.2)chr 16:g.71390658_71639890dup,片段大小:0.25 Mb。
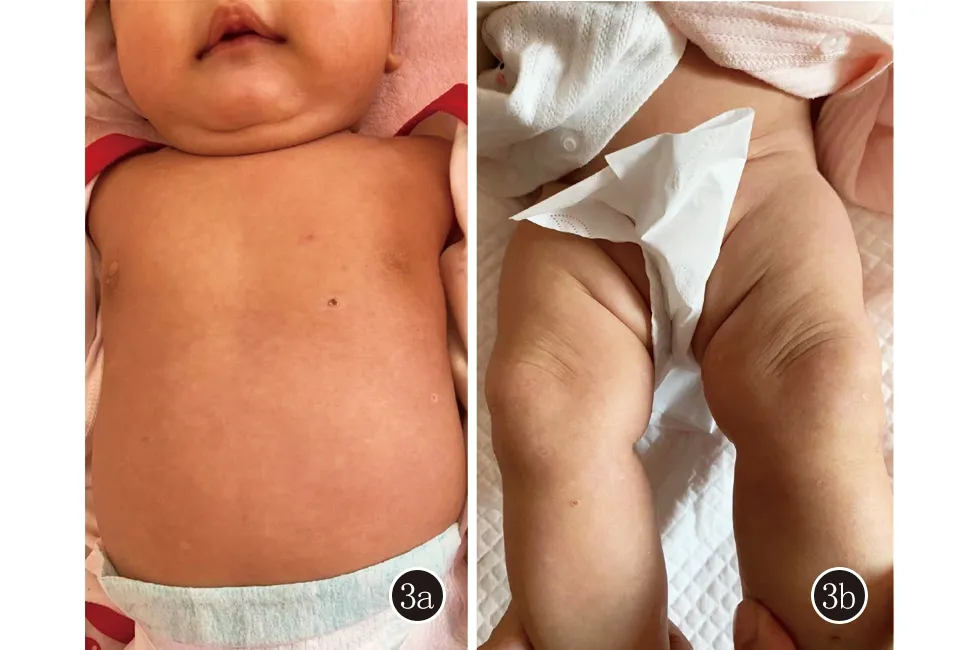
圖3 3個月軀干部(3a)、下肢(3b)皮損基本消退,無新發皮疹
討論先天性自愈性朗格漢斯組織細胞增生癥臨床表現具有多形性,可表現為丘疹、結節、結痂、水皰,較少出現出血性大皰,有時出現潰瘍,甚至軟疣樣丘疹。1973年Hashimoto和Pritzker首次報道[1],目前有文獻報道100余例, 國內文獻報道該病共6例。皮疹通常發生在出生時或在新生兒期,大多數患者無系統性損害,但也有少數病例報道有伴發肺和眼部損害[2]。經數月后皮損開始自行消退,所有報道的病例都在1年內痊愈。本病能自愈,可不必治療。但如果皮疹持續,可局部使用糖皮質激素、他克莫司或氮芥,皮疹局限者可手術切除[3]。
皮膚共聚焦顯微鏡(RCM)示大量中度折光的炎細胞浸潤,同部位病理取材,行組織病理、免疫組化S100、CDla染色,證實明亮的樹突狀細胞為活化的朗格漢斯細胞,對比RCM圖像與組織病理、免疫組化病理S100、CDla結果具有一致性[4]。CD1a是LC的一種分化性抗原,具有持續、穩定表達的特性,它被認為是鑒定人外周血與骨髓樹突狀細胞的最佳選擇,也是確診LCH的最佳標志物。Langerin是LC特異性凝聚素,它的靈敏度和特異度均等同于CD1a[5]。
組織病理和免疫組化仍是診斷朗格漢斯組織細胞增生癥(Langerhans cell histiocytosis,LCH)的金標準。自愈性和多系統性LCH在形態學和組織病理學上無法區分。不能僅根據實驗室檢測來區分它們。患兒3個月前出現病變和早期消退被認為更能預測是自愈性[6]。BRAF V600E突變患者比野生型BRAF患者表現出更高的疾病嚴重程度,一方面BRAF突變已被證實與內臟器官受累相關,另外研究發現BRAF突變與LCH復發之間存在統計學上的顯著相關性[7]。本例患兒BRAF V600E陰性,全外顯子測序陰性,更支持先天自愈性朗格漢斯細胞組織細胞增生癥的診斷。
