氣相色譜法測定胃痛片中4種化學成分含量*
2023-01-28 06:12:40古炳明鄔偉魁劉佳
中國藥業 2023年1期
古炳明,鄔偉魁,劉佳
(廣東省梅州市食品藥品監督檢驗所,廣東 梅州 514011)
胃痛片是由蒼術、厚樸、香附、高良姜、陳皮、碳酸氫鈉等制成的中西藥復方制劑,具有芳香行氣、和中止痛功效,用于胃酸過多、胃痛及脘悶、嘔吐等屬氣滯證的治療[1]。現行質量標準《國家食品藥品監督管理總局國家藥品標準胃痛片質量標準》WS-11084(ZD-1084)-2002-2012Z[2]僅對方中厚樸、碳酸氫鈉的含量進行測定,含量控制標準單一且未對方中君藥蒼術進行定量分析,不符合多組分、多靶點的現代中藥質量控制要求。蒼術為君藥,具有燥濕健脾、祛風散寒功效,主要活性成分蒼術素含量高低常用作評價蒼術的質量[3-4]。香附為臣藥,有調經止痛、理氣解郁功效,主要活性成分為α-香附酮[5]。本研究中建立了測定胃痛片中蒼術素、厚樸酚、厚樸酚和α-香附酮含量的氣相色譜法,可為其質量標準提升提供參考。現報道如下。
1 儀器與試藥
1.1 儀器
AB204-N型電子分析天平(瑞士梅特勒-托利多公司,精度為萬分之一);BP 211D型電子分析天平(德國賽多利斯公司,精度為十萬分之一);8890型氣相色譜儀(安捷倫公司);KQ-250DE型超聲波清洗器(昆山市超聲儀器有限公司,功率為300 W,頻率為50 kHz)。
1.2 試藥
蒼術素對照品(批號為111924-201806,含量以99.5%計),α-香附酮對照品(批號為110748-202016,含量以99.4%計),厚樸酚對照品(批號為110729-202015,含量以99.0%計),和厚樸酚對照品(批號為110730-201915,含量以99.8%計),均購于中國食品藥品檢定研究院;胃痛片(廣東嘉應制藥股份有限公司,批號分別為20180202,20190501,20200302);乙酸乙酯(廣州市番禺力強化工廠,批號為633379-05653)。
2 方法與結果
2.1 色譜條件
色譜柱:HP-5毛細管柱(30 m×0.32 mm,0.25μm),(5%)-二苯基(95%)-二甲基聚硅氧烷為固定相;程序升溫:初始溫度為50℃、保持4 min,以10℃/min的速率升至120℃、保持10 min,以10℃/min的速率升至220℃、保持10 min,以10℃的速率升至250℃、保持4 min;進樣口溫度:280℃;火焰離子化檢測器(FID)溫度:300℃;載氣:氮氣;流速:2.0 mL/min;進樣量:1 μL;分流比:1∶1。
2.2 溶液制備
取蒼術素對照品5.18 mg,α-香附酮對照品7.54 mg,分別置20 mL棕色容量瓶中,以乙酸乙酯溶解并定容;取厚樸酚對照品26.33 mg、和厚樸酚對照品25.81 mg,分別置10 mL容量瓶中,加乙酸乙酯溶解并定容,制成各對照品貯備液。分別取各對照品貯備液1 mL,置同一10 mL容量瓶中,加乙酸乙酯稀釋并定容,即得混合對照品溶液。取樣品,避光條件下操作,研細,取細粉2.0 g,精密稱定,置棕色容量瓶中,精密加入乙酸乙酯15 mL,稱定質量,超聲處理1 h,放冷后再次稱定質量,以乙酸乙酯補足減失的質量,搖勻,濾過,取續濾液,即得供試品溶液。根據處方工藝分別制備缺蒼術、香附、厚樸的陰性對照品,按供試品溶液制備方法制備陰性對照品溶液。
2.3 方法學考察
專屬性試驗:取2.2項下供試品溶液、混合對照品溶液及陰性對照品溶液各適量,按2.1項下色譜條件分別進樣測定,記錄色譜圖。供試品溶液中蒼術素、α-香附酮、厚樸酚、和厚樸酚色譜與混合對照品溶液色譜保留時間一致,各色譜峰分離良好,且陰性對照無干擾。色譜圖見圖1。
線性關系考察:分別精密吸取2.2項下各對照品貯備液0.60,0.80,1.00,1.20,1.40,1.60 mL,置同一10 mL容量瓶中,加乙酸乙酯稀釋并定容,按2.1項下色譜條件進樣測定,以峰面積(Y)為縱坐標、各成分的質量濃度(X,μg/mL)為橫坐標進行線性回歸。結果見表1。
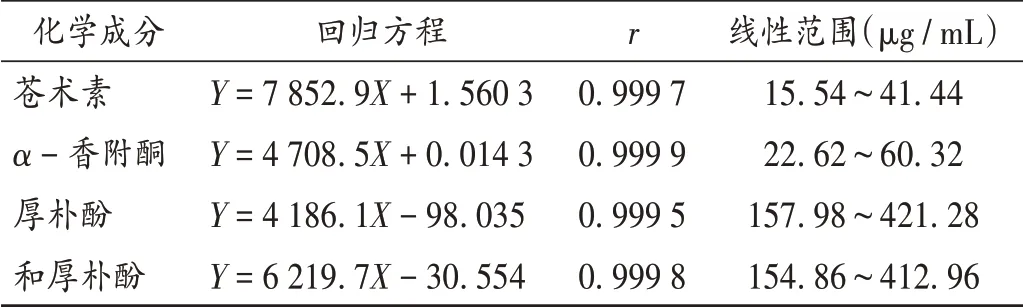
表1 線性關系考察結果(n=6)Tab.1 Results of the linear relation test(n=6)
精密度試驗:取2.2項下混合對照品溶液適量,按2.1項下色譜條件連續進樣測定6次,記錄色譜圖。結果蒼術素、α-香附酮、厚樸酚、和厚樸酚峰面積的RSD分別為0.32%,0.35%,0.52%,0.65%(n=6),表明儀器精密度良好。
重復性試驗:取樣品(批號為20180202)適量,共6份,依法制備供試品溶液,按2.1項下色譜條件分別進樣測定,記錄色譜圖。根據回歸方程計算得蒼術素、α-香附酮、厚樸酚、和厚樸酚的質量分數的RSD分別為1.08%,0.75%,0.81%,0.96%(n=6),表明方法重復性良好。

1.蒼術素2.α-香附酮3.厚樸酚4.和厚樸酚A.供試品溶液B.混合對照品溶液C-E.陰性對照品溶液(分別缺蒼術、香附、厚樸)圖1氣相色譜圖1.Atractylodin 2.α-Cyperone 3.Magnolol 4.HonokiolA.Test solution B.Mixed reference solution C-E.Negative reference solution(lacking Atractylodis Rhizoma,Cyperi Rhizoma and Magnoliae Officinalis Cortex,respectively)Fig.1 GC chromatograms
穩定性試驗:取樣品(批號為20180202)適量,按2.2項下方法制備供試品溶液,分別于0,2,4,8,24 h時按2.1項下色譜條件進樣測定,記錄色譜圖。結果蒼術素、α-香附酮、厚樸酚、和厚樸酚峰面積的RSD分別為1.25%,1.09%,1.13%,1.06%(n=5),表明供試品溶液24 h內穩定性良好。
加樣回收試驗:取樣品(批號為20180202)1.0 g,共6份,精密稱定,分別精密加入2.2項下混合對照品溶液5.0 mL,制備供試品溶液,按2.1項下色譜條件進樣測定,計算回收率。結果見表2。

表2 加樣回收試驗結果(n=6)Tab.2 Results of the recovery test(n=6)
2.4 樣品含量測定
取樣品(批號分別為20180202,20190501,20200302)適量,依法制備供試品溶液,按2.1項下色譜條件進樣測定,根據回歸方程計算蒼術素、α-香附酮、厚樸酚、和厚樸酚的含量,結果見表3。可見,各批樣品中成分含量存在差異,特別是未作規定的蒼術素和α-香附酮差異較大。

表3 胃痛片中4種成分含量測定結果(mg/g,n=6)Tab.3 Results of content determination of four constituents in Weitong Tablets(mg/g,n=6)
3 討論
3.1 色譜條件選擇
本研究中考察多種程序升溫的氣相色譜分離條件[6-8],預試驗中,最初程序升溫條件均設置進樣口溫度和檢測器溫度為300℃;程序升溫,以100℃保持7 min,再以20℃/min的速率升至250℃,保持10 min,分流比為20∶1。但在該條件下部分化學成分未能出峰,同時升溫速率過快導致主成分色譜峰過于集中,分離度差。更改條件后,進樣口溫度為280℃;檢測器溫度為300℃;程序升溫,初始溫度為50℃,保持4 min;以10℃/min的速率升至120℃,保持10 min;再以1.0℃/min的速率升至250℃,保持4 min;分流比為20∶1。但發現在該條件下色譜峰的響應值較低,調整分流比,1∶1時響應值在合理范圍內且各色譜峰峰形對稱,分離度符合要求,結果穩定,故選擇分流比為1∶1。
3.2 溶液制備操作
試驗初期,混合對照品溶液的部分色譜峰出峰時間和峰面積都不太穩定,特別是穩定性試驗中,放置時間越長,蒼術素和α-香附酮的色譜峰面積越小。查閱文獻[9-11],發現α-香附酮、蒼術素均為熱不穩定物質,且對光敏感、易氧化,故在對照品配置及供試品提取過程中,α-香附酮、蒼術素對照品及樣品均需避光保存。本研究中,供試品提取時間較長,為避免有效成分的分解失效導致檢測數據的不準確,超聲處理過程保持室溫,也可用冰袋控制溫度。
3.3 提取條件選擇
在制備供試品溶液和混合對照品溶液時,曾考察提取方法(超聲提取、超聲后離心提取)、提取溶劑種類(甲醇、乙醇、乙酸乙酯)、料液比及提取時間,發現以甲醇、乙醇作提取溶劑時,蒼術素和α-香附酮的色譜峰分離度很小,且受進樣時間長的影響,其他色譜峰不穩定,推測甲醇、乙醇的揮發性較大,導致目標成分濃度出現波動,采用乙酸乙酯為溶劑后解決了上述問題[12-14]。曾選擇α-蒎烯作為高良姜的定量指標,但后續研究發現無高良姜的陰性對照品中出現了α-蒎烯的干擾峰,查證文獻后得知香附揮發油中提取分離的單萜化合物有α-蒎烯、β-蒎烯、桉葉素、檸檬烯、γ-聚散花素和α-紫羅蘭酮等[15]。
3.4 方法評價
本研究中建立的方法操作簡便易行,精密度、重復性和準確度均良好,可為胃痛片的多成分含量測定和質量控制提供參考。
