印度梨形孢的熒光定量PCR檢測及其在水稻根系的定殖測定
2023-02-17 13:38:58李傳明韓光杰黃立鑫祁建杭陸玉榮
江蘇農業科學 2023年2期
夏 楊, 徐 彬, 李傳明,, 劉 琴,, 韓光杰, 黃立鑫, 祁建杭, 陸玉榮, 徐 健
(1.江蘇里下河地區農業科學研究所/國家農業微生物揚州觀測實驗站,江蘇揚州 225007; 2.揚州綠源生物化工有限公司,江蘇揚州 225008)
植物內生菌(endophyte)是一定階段或全部階段生活于健康植物的組織、器官內部或細胞間隙的真菌、細菌或放線菌[1]。內生菌長期定殖在植物體內,通過自身的代謝產物或借助于信號傳導作用于植物體產生影響,產生酶、激素、拮抗物質等來改善植物對非生物脅迫(干旱、鹽脅迫等)和生物脅迫(病原物、害蟲等)的耐受性[2]。梨形孢屬擔子菌門(Basidiomycota)層菌綱(Hymenomycetes)蠟殼耳目(Sebacinales)蠟殼耳科(Sebacinaceae)梨形孢屬(Piriformospora),是一類最早分化的擔子門菌根菌,長期定殖存活在植物根系。1998年首次從沙漠灌木根際分離獲得P.indica菌株[3],2012年在德國分離鑒定P.williamsii菌株[4]。大量研究證明,印度梨形孢寄主范圍廣泛,可以與200多種單子葉、雙子葉植物共生[5],通過增強植物對N、P等營養物質的吸收,促進植物生長,增強植物系統抗性,是一種具有廣泛應用潛力的多功能植物內生真菌[6-7]。
植物內生菌系統分布于植物的根、莖、葉、花、果實等器官和組織的細胞或細胞間隙,其種類、分布、定殖因植物種類及定殖部位不同而異[8]。不同于單細胞的內生細菌,印度梨形孢僅定殖于植物根系表面、表皮細胞和細胞間隙,并形成典型的梨形厚垣孢子,長期定殖存活在作物根系[9]。根系皮層梨形厚垣孢子的染色觀察成為檢測印度梨形孢在寄主植物上定殖共生的直接證據[10-12]。這種定性判別往往會受到取樣組織差異、雜質污染等外部因素影響。基于EF1α基因的PCR擴增檢測技術靈敏度較低,無法檢測出低拷貝模板DNA。綠色熒光蛋白標記法[13]可以對定殖部位和密度進行實時動態觀測,但此法必須以獲得綠色熒光蛋白標記突變株為前提且外源基因的引入可能會使其生物學效應降低。實時熒光定量PCR(quantitative real-time PCR,簡稱qPCR)作為一種快速、靈敏、準確的技術,已在病原體檢測[14]、基因表達量分析[15]、臨床疾病診斷[16]等方面得到了廣泛應用。本研究旨建立一種快速靈敏、準確高效的P.indica定殖定量的qPCR檢測方法,為深度開展印度梨形孢與寄主作物互作研究提供科學、準確的檢測方法。
1 材料與方法
1.1 試驗材料與處理
供試P.indica菌株PI-020由江蘇里下河地區農業科學研究所分離和保存。參考舒珊等的培養方法[17],取PI-020于PDA平板上活化,28 ℃培養5 d后,截取邊緣活性菌絲塊接種PDB培養液中,28 ℃、180 r/min振蕩培養7 d,勻漿后獲得孢子懸液。
供試水稻品種為南粳9108。2021年12月于室內將水稻種子漂洗、70%乙醇浸泡5 min、1%次氯酸鈉溶液浸泡5 min表面消毒后,無菌水沖洗3次并浸種1 d,28 ℃黑暗條件下催芽3 d。
挑選長勢一致的萌發種子置培養皿中,加入含PI-020(濃度為5×104CFU/mL)的營養液[18],以不含PI-020的營養液處理為對照組,于晝溫 30 ℃、夜溫26 ℃、光—暗周期14 h—10 h、相對濕度70%的條件下培養,每2 d更換1次營養液。分別于培養3、5、7、9、11、13 d取樣截取根部組織,每個時間點取樣10株,試驗設置3個重復。
1.2 試驗方法
1.2.1 DNA提取P.indica樣品準備:PI-020于PDA平板上進行活化,28 ℃培養5 d,刮取菌絲,液氮研磨;根系樣品準備:根系樣品10%甲醛浸泡 10 min,無菌水沖洗3次后晾干,液氮研磨。基因組DNA提取均采用CTAB(源葉生物,中國)法[19],采用NanoPhotometer N50(IMPLEN,德國)檢測DNA濃度,-20 ℃保存備用。
1.2.2 引物設計及質粒標準品的構建 根據P.indicaEF1α基因序列[20],參考Lin等的設計引物[21]Pi-F:5′-T C C G T C G C G C A C C A T T-3′,Pi-R:5′-A A A T C G C C C T C T T T C C A C A A-3′,利用NCBI的Primer-BLAST 工具完成對引物的特異性檢測。以P.indica基因組DNA為模板,進行PCR擴增,反應條件為:95 ℃ 3 min;95 ℃ 15 s,55 ℃ 15 s,72 ℃ 15 s,34個循環;72 ℃ 10 min。采用瓊脂糖凝膠DNA回收試劑盒回收PCR產物,并克隆至pCE2-TA/Blunt-Zero(諾唯贊,中國)載體上,獲得的重組質粒pCE2-Pi,作為qPCR擴增的質粒標準品(濃度為90 ng/μL),根據質粒拷貝數計算公式,換算成拷貝數為2.03×1010copies/μL。
1.2.3 qPCR標準曲線的建立及靈敏度檢測 對上述質粒標準品pCE2-Pi進行10倍梯度稀釋,備用。利用StepOnePlus實時熒光定量PCR儀(ABI,美國),以2.03×107~2.03×102copies/μL共6個濃度梯度的pCE2-Pi質粒標準品為模板進行qPCR擴增。反應體系為2×ChamQ SYBR qPCR Green Master Mix 10 μL,50×ROX Reference Dye 1 0.4 μL,上下游引物(10 μmol/L)各0.4 μL,模板DNA 1 μL,ddH2O 7.8 μL。反應條件為:95 ℃ 30 s,95 ℃ 10 s,60 ℃ 30 s,40個循環。以標準品拷貝數的對數為橫坐標,CT值為縱坐標,建立標準曲線。同時進行常規PCR擴增,比較qPCR檢測的靈敏度。
1.2.4 水稻苗根系P.indica的定殖檢測 參考袁聽等的臺盼藍染色法[10]對定殖培養水稻根系樣品進行染色、鏡檢觀察。同時,以無菌水作為稀釋液,將提取的水稻根系樣品DNA稀釋至相同濃度(50 ng/μL),利用常規PCR法[21]以及上述已建立的qPCR方法,對樣品DNA進行檢測。
1.3 數據統計與分析
采用SPSS 22軟件對試驗數據進行單因素方差分析(one-way ANOVA)和F檢驗(Tukey’s檢驗),利用Microsoft Excel 2019、GraphPad Prism 8軟件進行圖表繪制。
2 結果與分析
2.1 引物檢測及質粒標準品的制備
基于P.indicaEF1α基因的序列,通過引物 Pi-F/Pi-R對P.indica基因組DNA進行PCR擴增,獲得了大小為84 bp的目的片段(圖1)。利用該引物對實驗室常備材料水稻、枯草芽孢桿菌(Bacillussubtilis)、蘇云金芽孢桿菌(Bacillusthuringiensis)、灰梨孢(Pyriculariaoryae)、婁徹氏鏈霉菌(Streptomycesrochei)等DNA進行擴增,均未檢測到目標條帶(圖略),表明所設計的引物特異性較好。目的片段經切膠回收,連接至pCE2-TA/Blunt-Zero載體,并轉化大腸桿菌感受態細胞Trans1-T1,所得轉化子通過菌液PCR鑒定,電泳獲得大小為250 bp的明亮單一條帶(圖2),符合預期大小。目的片段經測序鑒定正確,表明重組質粒構建成功。

2.2 qPCR標準曲線的建立及靈敏度分析
以2.03×107~2.03×102copies/μL共6個濃度梯度的質粒標準品作為模板進行qPCR擴增,獲得不同濃度質粒標準品的擴增曲線(圖3-A),當質粒標準品濃度為2.03×102copies/μL時, 仍有擴增曲線。常規PCR結果表明,質粒標準品濃度為2.03×107~2.03×105copies/μL時,電泳有明顯條帶。當質粒標準品濃度小于2.03×105copies/μL時,電泳無條帶(圖3-B),檢測靈敏度為2.03×105copies/μL。說明建立的qPCR法檢測靈敏度能達到2.03×102copies/μL,比常規PCR高1 000倍。根據qPCR擴增結果,獲得不同起始模板所對應的各自CT值。以起始模板對數為X軸、CT值為Y軸,所建立的標準曲線表明,隨著樣品濃度的減小,CT值逐漸增加,兩者呈線性關系,標準曲線決定系數r2=0.999,說明擬合的線性回歸方程效果較好。所得標準曲線的線性回歸方程為y=-3.945x+42.045(圖4)。

2.3 水稻苗根系P. indica定殖檢測
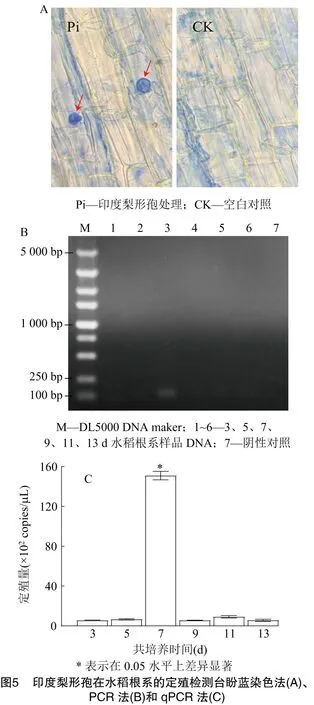
以染色法、常規PCR法、qPCR方法檢測水稻苗根系P.indica的定殖,比較不同檢測方法的差異。臺盼藍染色鏡檢P.indica處理水稻根系,僅在處理7 d的樣品部分根系直接觀察到經染色后呈深藍色的厚垣孢子,對照未發現P.indica孢子定殖,說明染色鏡檢受取樣等因素影響大,不能準確反映P.indica的實際定殖情況(圖5-A)。常規PCR檢測同樣是處理7 d的水稻根系樣品DNA能擴增出 84 bp 的目的條帶,其余DNA均未擴增出條帶(圖5-B)。采用qPCR法對水培水稻苗根系P.indica定殖量進行檢測,所有樣本中均能檢測到P.indica,P.indica處理3 d即成功定殖于水稻苗根系,定殖量為5.54×102copies/μL,5 d上升至 6.62×102copies/μL,7 d顯著上升,達到最高定殖量1.51×104copies/μL(DNA),隨后定殖量顯著下降,9 d定殖量為5.58×102copies/μL,13 d仍能檢測到5.58×102copies/μL的定殖量(圖5-C)。


3 討論
P.indica在植物根系穩定定殖是其發揮功能多樣性的必要條件,開展高效精準的P.indica定殖檢測是P.indica基礎研究工作中的重要環節。qPCR具有精準定量、高效靈敏等優點,可用于樣品中特定DNA序列的定性定量分析。李磊等基于立枯絲核菌融合群AG3的TEF保守區間設計特異性引物,建立的立枯絲核菌qPCR檢測體系靈敏度為19.5 fg/μL,是常規PCR的1 000倍[22]。Dubey等基于ITS序列同源性設計引物,用于番茄病原菌(立枯絲核菌)的檢測,其中常規PCR法檢測下限為0.025 ng/μL,而qPCR法檢測下限為1.24 pg/μL,靈敏度遠高于常規PCR法[23]。傳統的臺盼藍染色法雖然簡單、快捷、直觀、方便,目前作為主要的檢測方法應用于P.indica定殖能力的檢測試驗[24-25],但在進行P.indica定殖檢測時,其結果的準確度往往受根系取樣部位所影響,同時只能局部顯微鏡檢,不能全面反映內生菌的定殖情況,影響了試驗的準確性。盛萍萍等研究表明,染色前樣品根系的處理還應控制好KOH溶液濃度和處理時間,主要由寄主作物種類及其木質化程度所決定[26]。對于木質化程度較低的草本植物幼嫩根系,過高濃度(>10%)的KOH溶液和過長(>60 min)的處理時間都會對根系皮層細胞結構造成破壞,染色后難以觀察和區分。因此,應根據P.indica自身定殖特點結合寄主作物種類,選擇合適的部位(根系成熟區),運用恰當的處理方式進行檢測,同時增加取樣范圍和頻次,以進一步提高檢測結果的準確度。常規PCR法雖然具有較高的靈敏度,但對于根系表面定殖數量相對較少的內生菌而言,仍難以檢測到低拷貝數的DNA樣品,同時不能準確統計定殖數量,分析定殖效應。本研究所建立的P.indicaqPCR定殖定量檢測方法具有較高的擴增特異性,能呈現出良好的“S”形擴增曲線,基于起始模板對數與CT值所建立的標準曲線線性關系良好,靈敏度能達到2.03×102copies/μL,比常規PCR提高1 000倍,因此可以很好地用于P.indica的定殖定量檢測。
4 結論
本研究建立了能夠快速、精準、高效地對水稻根系P.indica定殖進行定量檢測的qPCR方法,分析該方法相比于臺盼藍染色法和常規PCR法在準確度和靈敏度上所表現出來的優越性,為深度開展印度梨形孢與寄主作物互作研究提供科學、準確的檢測方法。
猜你喜歡
青少年科技博覽(中學版)(2022年6期)2022-12-27 19:44:27
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
軍事文摘(2021年22期)2021-11-26 00:43:51
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
中學生數理化·七年級數學人教版(2021年6期)2021-11-22 07:50:58
文苑(2020年6期)2020-06-22 08:41:52
文苑(2019年22期)2019-12-07 05:29:00
當代陜西(2019年8期)2019-05-09 02:22:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
