紅木家具涂飾用漆蠟鉛的不確定度分析
2023-02-21 10:27:12許超群
山西化工 2023年1期
關鍵詞:標準
許超群
(國家木雕及紅木制品質量監督檢驗中心(浙江),浙江 東陽 322100)
紅木家具不僅實用性強,而且滿足消費者環保和耐用性要求。隨著“綠色消費”理念日益普及,其裝飾的安全功能也日漸重視。漆蠟常用作紅木家具涂飾輔料,其中重金屬元素含量備受關注。根據國家強制性標準GB 28010—2011《紅木家具通用技術條件》[1]本文以微波消解—石墨爐原子吸收光譜法測定漆蠟中鉛含量為例,按照《測量不確定度評定與表示(JJF 1059.1—2012)》[2]中規定,探討方法不確定度。
1 實驗部分
1.1 儀器
原子吸收光譜儀,240ZAA,美國安捷倫科技有限公司;微波消解儀,WX-8000,上海屹堯儀器科技發展有限公司;加熱板,DB-3,常州國華電器有限公司;電子天平,CPA225D,德國賽多利斯;可調移液器,上海精宏實驗設備有限公司。
1.2 主要試劑
鉛標準液,500 μg/mL,編號為GSB 07—1282—2000,環境保護部標準樣品研究所;硝酸,優級純;純水,GB/T 6682 規定的二級水。
2 實驗方法與過程
2.1 取樣、稱量與前處理
用刀片切去漆蠟表面氧化部分,并通過磁力攪拌器粉碎。稱取0.2 g~0.8 g 左右(精確至0.001 g)漆蠟樣品放到微波消解瓶中,加入5 mL 硝酸,參照微波消解程序(表1)將樣品消解。待消解完成后冷卻至室溫取出,將消解罐在卻140 ℃~160 ℃電熱板趕酸至1 mL左右。待消解罐冷卻后,將消化液轉移至25 mL 容量瓶中,用少量水洗滌消解罐2 次~3 次,合并洗滌液于容量瓶中并用水定容至刻度混勻。同時做試劑空白實驗。

表1 微波消解程序表
2.2 標液配制
準確吸取鉛標準溶液(500 μg/mL)1.00 mL 于100 mL 容量瓶,加硝酸溶液(5+95)至刻度,混勻得鉛一級標準中間液(5 mg/L),再用1.00 mL 單刻度移液管吸取一級標準溶液于100 mL 量瓶中定容,混勻得二級鉛標準中間液(50 ng/mL)。
3 建立數學模型
參照《食品安全國家標準食品中鉛的測定》(GB 5009.12—2017)中規定的方法,漆蠟樣品經微波消解完全后,對其鉛的殘留量建立數學模型,見式(1):

式中:Q 為漆蠟中鉛的含量,mg/kg;ρ 為樣品溶液中鉛的質量濃度,μg/mL;ρ0為空白溶液中鉛質量的濃度,μg/mL;V 為試樣消化液定容體積,mL;m 為試樣質量,g。
4 不確定度來源分析
根據數學模型分析不確定度來源,如圖1 中可以看出,紅木家具涂飾用漆蠟中鉛測量不確定度主要由漆蠟樣品稱量(m)、試樣消解液定容(V)、前處理(p)以及鉛質量濃度(ρ)組成。可表示為式(2):


圖1 不確定度來源分析
5 測量不確定度分量的評定[3]
5.1 漆蠟樣品稱量引入的相對標準不確定度urel(m)
用電子分析天平稱量,稱取漆蠟質量為0.501 0 g,根據檢定證書最大允差為±0.5 mg,按均勻分布,k=,由于稱量時有去皮和稱重兩次操作,分量應計算兩次,則:,所以urel(m)=
5.2 消解液定容引入的相對標準不確定度ur(V)
5.2.1 容量瓶允差引入的標準不確定度u1(V)
消解液定容采用A 級容量瓶為25 mL,允差為±0.030 mL,按三角形分布,,則:u1(V)=
5.2.2 溫度變化引入的標準不確定度u2(V)
假定實驗室環境溫度變化在20 ℃±5 ℃,水的體積膨脹明顯大于容量瓶的體積膨脹,考慮水的體積膨脹系數為2.1×10-4/℃,按均勻分布,則:u2(V)=
5.2.3 估讀誤差引入的標準不確定度u3(V)
單標線25 mL 容量瓶充滿液體時,估讀誤差約為±0.01 mL,按均勻分布,0.005 8。
5.3 鉛質量濃度ρ 引入的相對標準不確定度urel(ρ)
5.3.1 標準溶液配制過程引入的相對不確定度
鉛標準溶液證書(編號GSB 07—1282—2000)中提供相對擴展標準不確定度(k=2)為1%,得urel(ρ1)=0.005。
5.3.2 配制標準溶液引起的相對標準不確定
稀釋過程:準確吸取鉛標準溶液(500 μg/mL)1.00 mL 于100mL 容量瓶,加硝酸溶液(5+95)至刻度,混勻得一級鉛標準中間液(5 μg/mL)。再用1.00 mL單刻度移液管吸取鉛一級標準溶液于100 mL 量瓶中加硝酸溶液(5+95)至刻度定容,混勻得二級鉛標準中間液(50 ng/mL)。
查閱A 級1 mL 移液管最大容許誤差為0.007 mL和A 級100mL 容量瓶的最大容許誤差為0.10 mL,A級5 mL 移液管最大允許誤差為0.015 mL,均采用均勻分布[4]。

5.3.3 標準曲線擬合引入的相對不確定度
標準溶液吸光度測定數據如表2 所示,曲線擬合得到一元線性方程為Abs=0.002 19×ρ+0.006 66(R=0.999 3)。

表2 鉛標準溶液濃度與吸光度
由標準曲線擬合引起的不確定度計算公式,見式(3)、式(4)[5]:
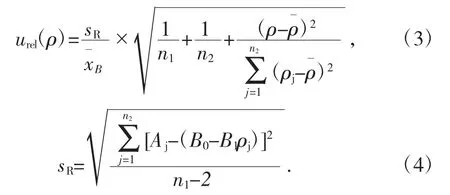
式中:n1為被測溶液測量次數;n2為5 個標準溶液總共測量次數;ρj為標準溶液的質量濃度值;ρˉ為標準溶液質量濃度平均值;Aj為標準溶液的吸收率測定值;B1為標準曲線斜率;B0標準曲線的截距。
將表2 中數據代入式(4),得到sR=0.001 268。擬合標準曲線所引入的相對標準不確定度為:urel(ρ3)=0.045 6。
5.3.4 重復性測定引入的相對標準不確定度
取同一份漆蠟樣品測定6 次,結果如表3。
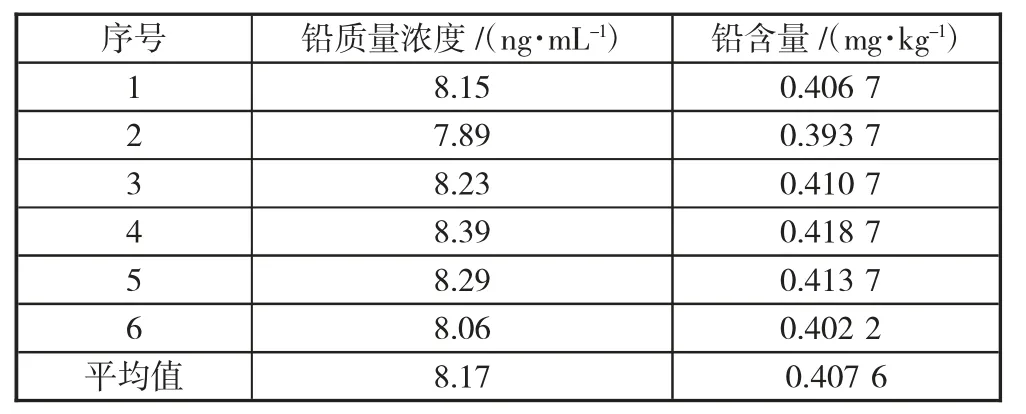
表3 樣品中鉛含量的測定結果
由貝塞爾公式(5)[5]可得:
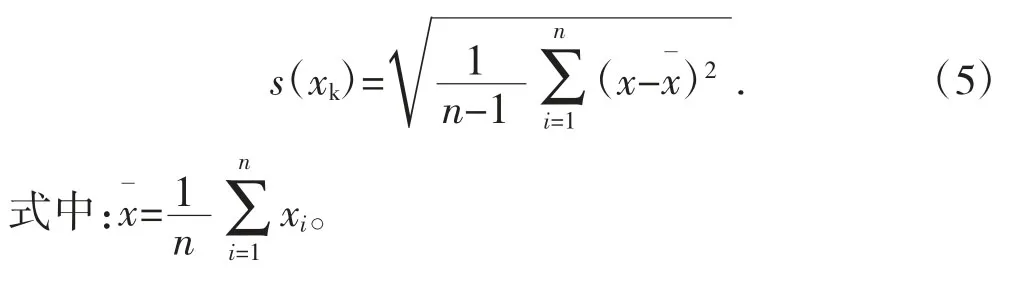
計算鉛含量的標準偏差s=0.008 85,則由于重復性導致A 類標準不確定度為
5.4 前處理引入的相對標準不確定度
前處理主要通過微波消解回收率引入的不確定度,結合本實驗室數據,本方法鉛回收率范圍在92%~109%,采用近似公式(6)計算:
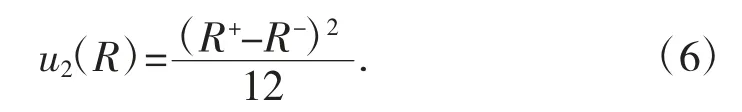
式中:u(R)為回收率不確定度;R+為回收率的上界;R-為回收率的下界。經計算u2(R)=0.002 4。
5.5 擴展不確定度及報告
漆蠟中鉛含量的合成相對標準不確定度計算為式(7):
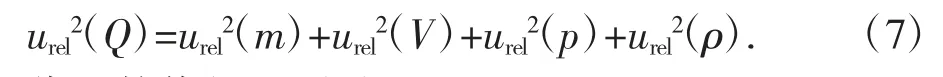
代入數值得:urel(Q)=0.046 9。
合成標準不確定度:u(Q)=0.046 9×0.407 6=0.019 mg/kg。
按正態分布,在置信概率95%時選擇包含因子k=2,則紅木家具涂飾用漆蠟鉛含量測定結果的擴展不確定度報告為0.4076 mg/kg±0.038 mg/kg(k=2)。
6 結果與討論
本文通過微波消解—石墨爐原子吸收光譜法對漆蠟中鉛含量進行不確定度評價,從評定結果看,不確定度主要由標準曲線擬合所引入。因此,在實驗檢測過程中,增加平行測定次數,并嚴格按規范進行操作,盡可能降低不確定度,提高實驗室的檢測水平。
猜你喜歡
城市道橋與防洪(2022年4期)2022-07-01 06:04:12
當代陜西(2019年8期)2019-05-09 02:22:48
上海建材(2019年1期)2019-04-25 06:30:48
動漫星空(興趣百科)(2019年3期)2019-03-07 07:23:10
家庭影院技術(2018年4期)2018-05-09 07:07:52
專用汽車(2016年4期)2016-03-01 04:13:43
質量與標準化(2015年9期)2015-12-31 11:41:40
中國質量與標準導報(2014年4期)2014-03-11 19:54:25
中國質量與標準導報(2014年10期)2014-02-28 22:25:47
中國質量與標準導報(2014年7期)2014-02-28 22:24:39
