核殼結構在甲烷干重整中的應用
2023-03-01 07:40:10鄧少碧邊洲峰
化工進展 2023年1期
鄧少碧,邊洲峰
(東南大學能源與環境學院,江蘇 南京 210096)
隨著現代工業和社會活動的發展,化石燃料的消耗不斷增加,同時釋放出大量的二氧化碳,造成溫室效應、氣候異常等問題,因此對二氧化碳的捕集和利用已成為一個重要且緊迫的課題[1]。甲烷與二氧化碳的重整又稱甲烷干重整,可以將兩種溫室氣體(CO2和CH4)轉化為合成氣(CH4+CO2→2H2+2CO,ΔH=247kJ/mol),生成的合成氣又可用于生產各種化學品和燃料[2?3],目前已受到學術界的高度關注。
對于甲烷干重整的熱催化反應,CO2和CH4具有穩定的化學性質[4],加之該反應的強吸熱特性,通常需要高溫(700℃以上)才能實現反應。貴金屬(Ru、Rh、Pt等)和Ⅷ族(Fe、Co、Ni)金屬均具有甲烷干重整的催化活性[5?6]。貴金屬比鎳基催化劑具有更高的活性和更好的耐積炭性,但貴金屬儲量少、價格高,而過渡金屬鎳來源廣泛、價格較低,所以綜合考慮,鎳被認為是甲烷干重整的首選工業催化劑。然而,在甲烷干重整反應中,鎳等活性金屬在高溫下會發生遷移和團聚,表面有效活性位點數目減少,活性下降[7]。另一方面,重整反應往往伴隨一些副反應,主要是甲烷的裂解(CH4→C+2H2,ΔH=75kJ/mol)和一氧化碳歧化(2CO→C+CO2,ΔH=?172kJ/mol),這兩個副反應會生成碳,進而沉積在催化劑的表面,造成失活。由于活性金屬顆粒的燒結和在金屬表面積炭等原因,甲烷干重整催化劑的穩定性較差,這也限制了其工業應用[8]。為解決甲烷干重整中催化劑活性金屬燒結和碳沉積的問題,國內外學者開展了大量研究,比如通過增強金屬和載體的相互作用來增加催化劑的穩定性和活性[9?10];開發高度分散的Ni?M(M=Sn、Co、Au、Pt 等)雙金屬催化劑,利用雙金屬協同作用提高抗積炭性能[11?12];通過摻雜堿性或稀土金屬來改變載體性質[13?14]等。
近年來,核殼結構的出現為甲烷干重整催化劑的開發提供了新的思路。核殼結構由核與殼組成,一般情況下,核為活性金屬顆粒,殼為多孔殼層,以便反應物和產物的進出。核殼結構的空間限域效應是其優勢的本質原因:由于殼的存在將活性金屬限制在固定的空間范圍內,從而避免燒結;同時也阻斷了絲狀碳的生長空間,抑制了積炭的生成。因此,核殼結構已成為研究熱點,Das 等[15]總結綜述了核殼結構在CO2轉化利用中的應用,包括熱催化、光催化和電催化。而蔡雨露等[16]綜述了鎳基核殼結構催化劑的制備及其在甲烷干重整中的應用,著重介紹了核殼結構的多種制備方法。
目前,研究者們對核殼結構在甲烷干重整中的應用進行了更精細化的研究。①對核殼結構進一步拓展,從最基本的單核單殼結構到多核同殼結構,基于核殼結構的蛋黃殼結構、三明治型核殼結構也被研究者們開發出來。②對內核進行進一步的設計:核既可以是Ni、Co、Ru 等單金屬,也可以是Ni?Co、Ni?In等雙金屬合金,還可以是含有活性金屬的復合納米材料。③對殼層進行進一步的研究:因為高熱穩定性、高比表面積、可調節孔徑分布和易于合成等原因,SiO2是最常用的殼層材料。Al2O3也是常用殼層材料,因為其具有堿性。除此以外,一些金屬氧化物,如CeO2、ZrO2等也是殼層材料的選擇,具有多孔結構的沸石也被作為殼層材料研究。本文根據殼層材料的不同,將核殼催化劑大致分為氧化硅、氧化鋁和其他殼層三類,并從制備方法、形貌結構、催化特性的角度分別進行了系統地綜述,最后展望了核殼結構在未來的研究方向。
1 核殼結構在甲烷干重整中的應用
1.1 氧化硅殼層
二氧化硅由于具有高熱穩定性、高比表面積,可調節孔徑分布和易于合成,是目前應用最廣泛的殼層。研究者們一般通過正硅酸乙酯(TEOS)水解制備氧化硅殼層。對于氧化硅殼層的研究現狀,一方面,研究者們分析不同的核的催化劑性能,如最常見的Ni[17?18]以及Co[19]、Ru[20]等;同時也可以以合金為核,如Ni?Co 合金[21]、In?Ni 合金為核[22]等。另一方面,氧化硅殼層厚度和孔隙率易調節,殼的厚度和孔隙率對催化反應的影響研究是常見課題。
Zhang 等[17]通過溶膠凝膠法合成了Ni@SiO2催化劑,并在甲烷干重整反應中進行實驗,在反應溫度高達850℃時,催化劑依舊被證明是熱穩定的,而且多孔二氧化硅外殼顯著抑制了燒結和結焦。Peng等[18]對Ni@SiO2結構進行進一步改造,采用反相微乳液法制備出多核同殼(M?Ni@SiO2)催化劑。在甲烷干重整的測試中,20h 后,M?Ni@SiO2對CH4和CO2的轉化率沒有明顯的降低,而傳統負載型催化劑Ni/SiO2和Ni/Al2O3的轉化率大幅下降。除了單核單殼結構外,Bian等[23]對核殼結構進行了進一步拓展,合成了三明治型核殼結構SiO2@Ni@SiO2(圖1),該催化劑合成過程不需要任何有毒的有機溶劑,且在600℃低溫DRM中表現出優異的催化性能,幾乎沒有積炭生成。
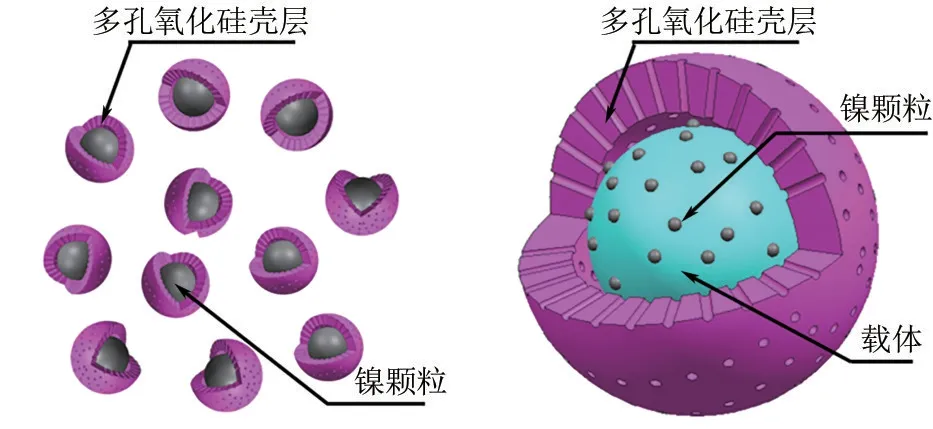
圖1 單核單殼和三明治核殼結構示意圖
研究者們探究了更多可能的金屬@SiO2結構中核的材料選擇。Pang等[19]制造了以Co為核的x?Co@SiO2?y(x為Co的粒徑,nm;y為SiO2的殼層厚度,nm)催化劑,通過優化核殼尺寸、殼層厚度和核殼相互作用,27.8?Co@SiO2?14.3 表現出最優的活性和穩定性。Yang等[20]制備了Ru@SiO2催化劑,通過優化參數,0.6% 800c?40p?2s 催化劑性能較佳(Ru 負載量為0.6%,煅燒溫度為800℃,催化劑制備溫度為40℃,正硅酸甲酯含量為2mL)。與傳統負載型催化劑0.6% Ru/SiO2和無載體Ru 催化劑相比,核殼催化劑具有最高的初始活性和最長的壽命。
上述單金屬為核,氧化硅為殼的催化劑,均表現出優良的穩定性和活性。以合金為核,利用雙金屬協同作用,可以進一步改變催化劑的性能,如核中摻雜CeO2可以提供氧空位,有助于CO2活化等。Zhao 等[21]通過微乳液法合成NiCo@SiO2,該催化劑表現出較高的活性和選擇性,并且在800℃的甲烷干重整反應中表現出了很長的使用壽命(>1000h)。同時NiCo@SiO2催化劑活性遠遠高于以單金屬為核的Ni@SiO2和Co@SiO2催化劑。Bian等[24]以Ni?Mg層狀硅酸鹽納米管為核,覆蓋SiO2殼層,經H2還原后Ni 納米顆粒支撐在納米管上,同時被氧化硅殼層限制(圖2),該催化劑在750℃的72h 試驗中表現出良好的穩定性和優異的抗積炭性能。
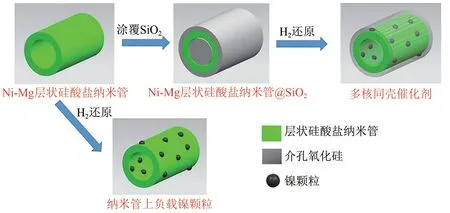
圖2 由Ni?Mg PSNTS@SiO2衍生的多核同殼催化劑的制備方案[24]
Zhao 等[25]在核殼結構中摻雜金屬氧化物CeO2,形成NiCe@m?SiO2結構。核由Ni 和CeO2組成,位于二氧化硅殼內。由于CeO2的加入,二氧化硅表面產生更多的活性氧物種并改善了鎳納米顆粒的分散性,從而提高催化劑性能和抑制碳沉積。Liu等[22]合成了InxNi@SiO2催化劑。結果表明,In的電負性較小,電子從In 上轉移到Ni,電子云密度的增加削弱了Ni 激活C—H 鍵的能力,減少CH4的裂解,進而減少C 的沉積,但CH4的轉化率相比Ni@SiO2催化劑會有所降低。
研究者們對影響核殼結構催化劑活性和穩定性的因素也進行了研究。Li 等[26]研究殼層厚度對Ni@SiO2的影響,SiO2殼層為3.3nm 時,由于Ni 與SiO2殼層相互作用力弱,導致催化劑不穩定,Ni嚴重燒結,碳大量沉積,殼層增加至5.7nm,穩定性顯著提高,而當殼層厚度大于8.6nm時,Ni與SiO2殼層界面會形成層狀硅酸鎳物質,在還原后會分解成小的鎳顆粒,提高了催化劑的活性;殼層厚度為11.2nm時,催化劑活性達到最高,且此時核殼結構變為蛋黃殼結構。Pang等[27]研究孔隙率對RuCo@SiO2的影響,采用兩種表面活性劑(CTAB 和PVP)分別改變孔隙率,研究表明較高的孔隙率可以提高傳質效應,進而增加催化劑的宏觀活性。但當溫度大于700℃時,殼層孔隙率的影響不太顯著。
Wang 等[28]發現殼的擴散效應(主要是殼厚度和孔隙率)是影響核殼結構催化劑活性和穩定性的關鍵因素。殼的厚度和孔隙率通過改變反應物和產物的擴散路徑和擴散速度,進而影響催化劑的宏觀活性。此外,Wang 等進一步提出了模型解釋催化劑失活的原因。在該研究中,合成Ni@SiO2納米膠囊催化劑,通過加入不同TEOS 的量改變殼的厚度,將催化劑記為NSNC?Tx(x代表TEOS 加入的量,mL)。結果表明,流速為24L/(gcat·h)時,催化劑幾乎沒有失活,CO2和CH4的轉化率幾乎不變,而當流速變為96L/(gcat·h)時,催化劑明顯失活,但再次將流速變為24L/(gcat·h)時,NSNC-T10/25 活性會緩慢恢復,但NSNC?T5/20 活性只能部分恢復。據此,該作者認為“Gas wall”和“Hard blocker”模型(圖3)可解釋該現象。流速較高時,會形成氣壁,導致進出孔隙的流體速度減慢,甚至停止,造成“失活”,但這種“失活”是可逆的。而另一種失活原因是,水可能會引起氧化硅殼層孔結構的坍塌,阻礙反應物接觸活性位點,這種損壞對殼造成的傷害是永久性不可逆的。
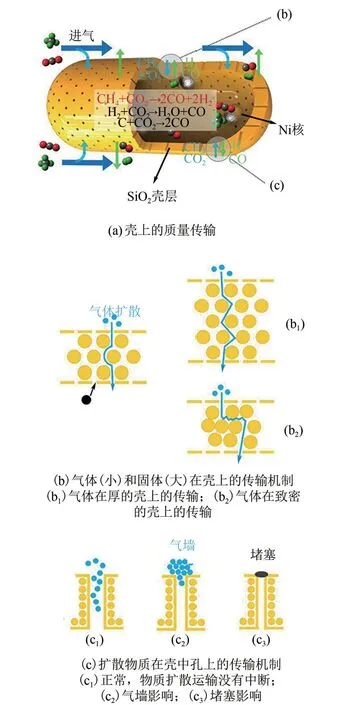
圖3 蛋黃殼NSNC?Tx催化劑的納米反應器工作原理示意圖,涉及殼擴散效應[28]
為了提高DRM反應的核殼結構催化劑性能,學者們還從增強傳熱的角度進行了研究。由于DRM反應的強吸熱性,核殼結構催化劑內部不可避免地會產生冷點。根據熱力學分析,這些冷點不僅降低了反應的轉化率,而且還促進了碳沉積,尤其是在鎳基催化劑上。Chai等[30]合成Ni@SiO2/Al2O3/FeCrAl?fiber催化劑。合金FeCrAl組成的超細纖維(FeCrAl?fiber)具有高溫耐受性、理想的機械穩定性、高滲透性、高傳熱性,可以增強傳熱,進而改善冷點問題。在800℃,氣體空間速度為5000 mL/(g·h)條件下,反應500h,Ni@SiO2/Al2O3/FeCrAl?fiber 催化劑沒有明顯的失活,同時幾乎沒有碳的沉積。
從現有的研究成果來看,SiO2作為殼層具有一定的優勢,主要是SiO2殼層的包覆采用成熟的Stober方法,制備過程簡單,同時殼層的厚度和孔隙率可以通過改變TEOS和表面活性劑的劑量實現調控。形成的氧化硅殼層是非晶態,具有很高的熱穩定性,所以適用于高溫下的反應。但是氧化硅殼層也存在一定的問題,主要是殼層帶來的傳質阻力,尤其是在殼層厚度較厚的時候,而氧化硅是惰性材料,對于重整沒有直接的促進作用,所以會在一定程度上降低表觀反應速率。
1.2 氧化鋁殼層
在DRM 傳統負載型催化劑中,Al2O3具有堿性,被認為是一種優良的載體材料,可促進CO2高效轉化[31]。相應地,國內外研究者們對于Al2O3能否作為核殼結構的殼層材料進行了探究。Huang等[32]將通過反相微乳液法合成的Ni@Al2O3催化劑和通過浸漬法合成的Ni/Al2O3催化劑在甲烷干重整反應中進行對比。Ni@Al2O3催化劑的鎳納米顆粒粒徑更小且活性表面積更大。同時,Ni@Al2O3表面形成大量的活性氧化物,更重要的是Ni@Al2O3也同樣能抑制鎳納米顆粒的遷移,避免燒結。
金屬@Al2O3催化劑通常通過反向微乳液法或原子層沉積法制備,而原子層沉積法可以很好地控制Al2O3殼層厚度。Baktash 等[33]采用原子層沉積法合成Ni@Al2O3。結果表明,覆蓋5個循環氧化鋁薄膜的催化劑(NiO?5催化劑)已經可以保護鎳顆粒在高達800℃的溫度下不燒結,產生高且穩定的甲烷轉化率。但是NiO?5 催化劑在低溫(525℃)下發生失活,主要原因是在較低溫度下發生了甲烷裂解和Boudouard反應,生成了積炭[34]。
Gould 等[35]將鎳納米顆粒使用原子層沉積法固定在球形氧化鋁載體上,在其上使用分子沉積法(MLD)覆蓋Al2O3薄膜,合成三明治結構的催化劑(圖4)。在973K 溫度下對該催化劑的活性和穩定性進行評估,結果表明經5次MLD循環的催化劑產生了最高DRM速率。但經過煅燒和還原后,5?MLD循環催化劑會失去部分活性,而10?MLD循環催化劑在反復煅燒和還原后也可保持原來的活性。可見在給定反應條件下,殼層厚度與活性和穩定性之間的平衡關系至關重要。
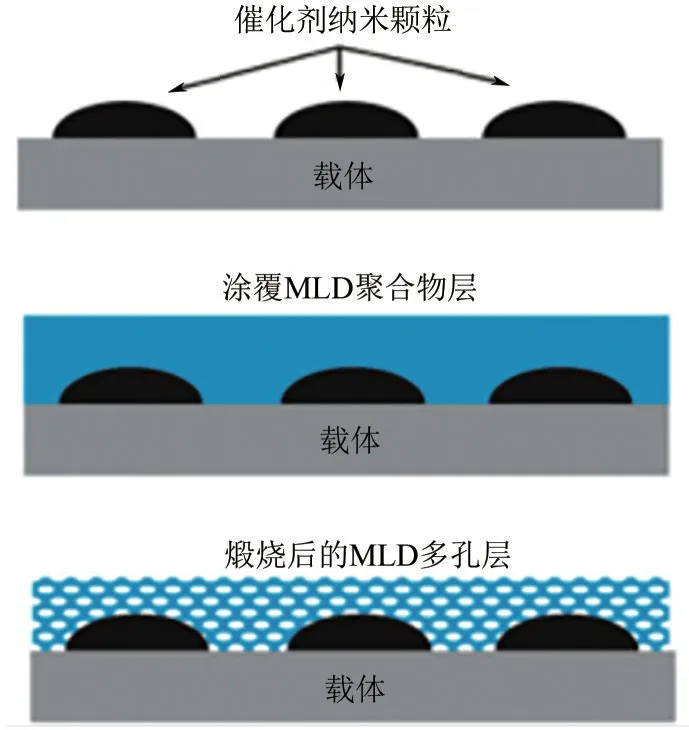
圖4 生產多孔氧化鋁覆蓋層的MLD涂層工藝示意圖[35]
Zhao等[36]同樣合成三明治型Al2O3/Ni/Al2O3催化劑,首先使用浸漬法將Ni 顆粒負載在Al2O3上,然后采用原子層沉積法覆蓋Al2O3薄膜。薄膜厚度對催化劑活性和穩定性影響很大,研究表明,80 層Al2O3薄膜的催化劑活性最高,CO2和CH4的轉化率都接近100%。在800℃的DRM 反應中可連續使用400h以上,且無明顯失活,具有良好的工業應用潛力。此外,Zhao 等對Ni 的負載量和煅燒溫度對催化劑性能影響進行了研究。結果顯示Ni 負載量為4%(質量分數),煅燒溫度為800℃時催化劑性能最佳。
Dai 等[37]制備了Ni/MgO@Al 催化劑(圖5),并與Ni/MgO 和Ni@Al2O3催化劑進行對比。結果表明MgO 核的加入可以增加鎳顆粒的分散性,同時發現雖然Ni/MgO@Al 和Ni@Al2O3都是屬于核殼結構,但沉積炭的物種不一樣,推測這與孔體積和孔徑不同有關。根據氮氣的物理吸附分析表明,Ni/MgO@Al 和Ni@Al2O3催化劑都具有較大的比表面積,這有助于提高反應氣體的傳質效率。
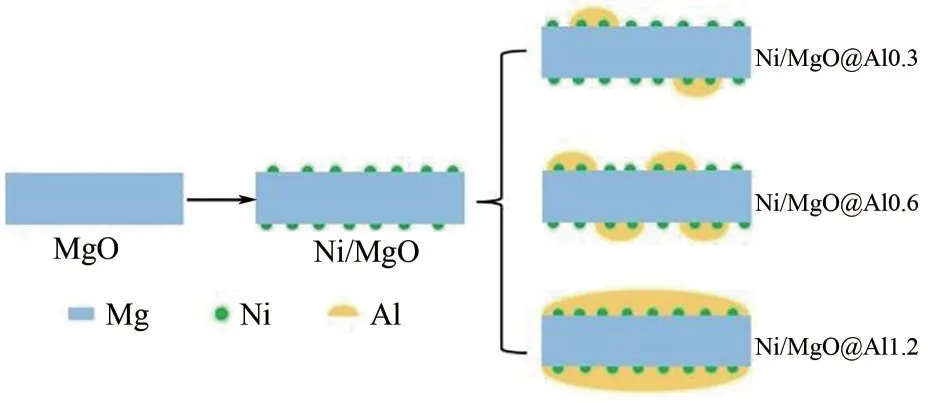
圖5 Ni/MgO@Alx(x=0.3,0.6,1.2)三明治結構催化劑示意圖[37]
Han等[38]比較了在Ni/SiO2上包覆SiO2和Al2O3殼層的兩種核殼結構催化劑在甲烷干重整中的活性,結果表明Al2O3殼層的催化劑的轉化頻率(TOF)是SiO2殼層的催化劑的大約3倍,原因是氧化鋁含有較多的堿性位點,增強了對于CO2的吸附作用,從而促進了反應。所以,相比較于惰性不參與反應的氧化硅,氧化鋁殼層的優勢在于它本身具有較多的堿性位點,可以促進反應的發生。而它的缺點也比較明顯,目前氧化鋁殼層的包覆大多采用原子沉積的方法,對于儀器的依賴度較高,增加了制備成本。此外,目前也沒有成熟的調控氧化鋁殼層孔隙率的方法。
1.3 其他殼層
除去SiO2和Al2O3作為殼層以外,國內外學者們也在積極探索其他可以作為殼層的材料。CeO2是一種常見的催化劑載體,具有豐富的活性晶格氧和氧空位。氧空位有助于CO2活化,移動氧能氣化碳前驅體,所以CeO2作為殼層材料已成為核殼結構催化劑的研究熱點。Tang等[39]通過以尿素為沉淀劑的水熱法合成了Ni@CeO2核殼催化劑,與催化劑Ni@SiO2進行對比。熱重分析結果表明,使用后的Ni@CeO2催化劑碳沉積量低于在Ni@SiO2上的碳沉積量。程序升溫氧化分析得到結果,Ni@CeO2上的碳主要是無定形碳(Cβ);Ni@SiO2上的碳主要是石墨碳(Cγ)。當CeO2作為殼層材料時,可以抑制沉積炭從無定型向石墨結構的轉變。Han等[40]制備雙殼結構Ni@SiO2@CeO2催化劑,CeO2的活性氧可以氣化碳前體,進而減少積炭。結果表明,相較于催化劑Ni@SiO2,Ni@SiO2@CeO2催化劑使甲烷干重整性能提高1.5 倍,且碳的沉積減小一半。Li 等[41]合成了Ni@NiPhy@CeO2催化劑。結果表明在氣體空間速度為36L/(gcat·h),溫度為700℃時,CH4和CO2的轉化率高且穩定,分別為72.8%、79.1%,表現出良好的性能。
ZrO2表面易產生氧空穴,可與活性組分相互作用,增強催化劑性能,熱穩定性能好,也是殼層材料的選擇之一[42]。Dou 等[43]采用濕化學合成路線研制了三明治型SiO2@Ni@ZrO2催化劑。多孔ZrO2殼層可以將SiO2核和多孔ZrO2殼層之間的Ni 顆粒穩定在6nm。該催化劑活性比未涂覆ZrO2殼層的催化劑高5~7倍,并且具有良好的抗積炭性。密度泛函理論分析表明,Ni15/ZrO2(SiO2@Ni@ZrO2的模型催化劑)不僅降低了CO2解離勢壘,而且有效地穩定了CH4/CO2重整反應中的反應物和中間體,如CH4、CO2、CHx(x=0~3)和CO,最終提高了CH4/CO2重整反應的催化活性和穩定性。
Han 等[38]比較不同金屬氧化物充當殼層材料的性能,Ni納米顆粒固定為5.2nm,在Ni/SiO2上覆蓋SiO2、Al2O3、MgO、TiO2、ZrO2五種不同的金屬氧化物殼層。結果表明,Al2O3、MgO 殼層催化劑的活性遠大于SiO2殼層催化劑,SiO2殼層催化劑活性近似于ZrO2殼層催化劑(圖6)。ZrO2具有較高的氧遷移率,能夠輕松去除沉積的碳物種,然而該實驗中ZrO2本身并沒有提高催化活性。TiO2殼層催化劑中,鎳納米顆粒嚴重聚集,造成催化劑低活性。
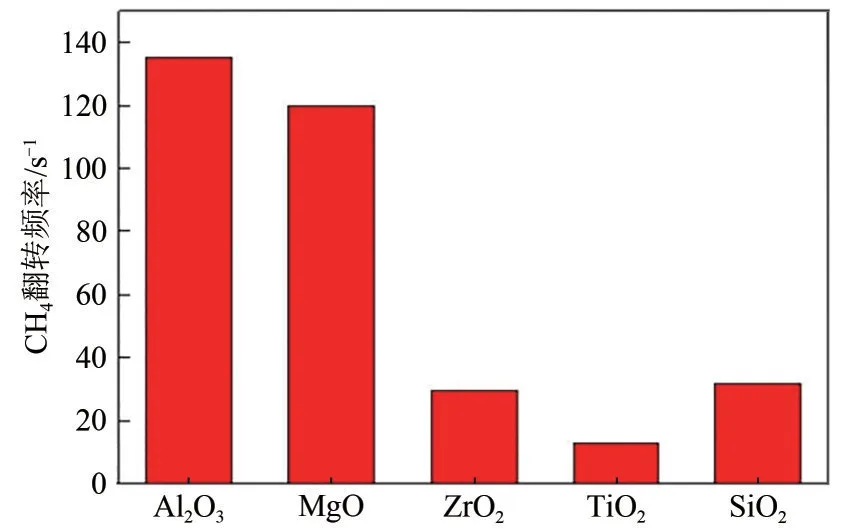
圖6 在Ni/SiO2上覆蓋金屬氧化物后CH4的轉化頻率[38]
除此以外,沸石也可以作為殼層材料。Dai等[44]采用空心沸石作為殼層材料,包覆Ni?Pt 雙金屬顆粒。研究表明,Ni 的存在可以顯著抑制Pt 的聚集和損失;Pt的存在可以顯著抑制Ni的燒結。1.5Ni?0.5Pt@Hol S?1 催化劑在甲烷干重整中反應6h 后,僅觀察到質量分數1.0%碳的沉積量。在制造1.5Ni@Hol S?1 催化劑時,殼層外會產生大的鎳顆粒,造成反應時碳的沉積,但由于殼層的保護,碳不會影響到殼層內小的鎳納米顆粒的活性。盡管產生了10.3%的積炭,1.5NiHol S?1 仍然具有較好的活性。
綜上所述,氧化鈰和氧化鋯也可以作為核殼結構催化劑的殼層材料。氧化鈰由于本身具有較多的活性晶格氧和氧空位,不僅有助于CO2的吸附,也能夠氣化沉積的碳物種,因此氧化鈰相比氧化硅來說,具有更高的催化活性和抗積炭性能。但是其缺點是目前氧化鈰殼層本質上是氧化鈰小晶體的堆疊,并不是完美包覆的殼層整體。同時,從目前關于氧化鋯的文獻報道來看,其是否能夠提高反應速率還存在分歧。此外,沸石也可以作為殼層材料,因為沸石具有更小的孔徑,可以更好地控制反應物和生成物的擴散,但是相比較于其他殼層材料,其制備過程略為復雜。
2 結語
相較于傳統負載型催化劑,核殼結構在甲烷干重整中的應用具有明顯的優勢,可以將活性金屬限制在殼層里,避免在高溫下發生金屬燒結,同時也控制了積炭的生成。所以說,核殼結構在干重整中的作用主要是通過空間限域效應增強催化劑的抗燒結和抗積炭能力,從而提高催化劑的穩定性。研究者們對不同的殼層材料進行了探索,應用最廣泛的是SiO2殼層,具有高熱穩定性,即使在高溫的甲烷干重整反應中也可保持結構的穩定,同時殼的厚度、孔隙率易調節,易于合成。其次是Al2O3殼層,除了限域作用以外,其本身具有較多的堿性位點,增強了對于CO2的吸附作用,從而促進了反應。CeO2殼層可產生氧空位,與活性組分相互作用,提高性能,所以也被作為殼層材料研究。沸石、TiO2等也可以為殼層材料,但在目前甲烷干重整反應中被研究的相對較少。多種殼層材料的相關研究都證明該核殼結構催化劑的優勢,但仍然存在一些問題需要進一步解決。
首先,殼層會覆蓋一部分的催化活性位點,減少了有效活性位點的數量,同時殼層的存在增加了反應物和產物往返于活性位點的擴散阻力。為了克服這一問題,可以對核殼結構進一步細化研究,構建更為先進的核殼結構。比如通過構建蛋黃殼結構,增加核殼之間的空心空間,降低傳質阻力,或者構建三明治的核殼結構,增強金屬?載體的相互作用,提高反應活性。其次,調控核殼結構的物理化學特性,比如殼厚度、孔隙率、金屬顆粒大小和形狀等,研究這些因素與催化活性之間的構效關系,為核殼結構催化劑的設計提供理論基礎。最后,在現有的可選的殼層材料之外,繼續對核殼材料進行探索,比如可以嘗試將SiO2、Al2O3、CeO2等殼層材料混合摻雜使用,優化殼層的性能。此外,目前核殼結構多用于甲烷重整,未來應該有在其他反應中的嘗試,比如乙醇重整、焦油重整,這些反應物分子更大,會對核殼結構的研究提出新的挑戰。
核殼結構在甲烷干重整工業化大規模應用也存在著亟待解決的問題。雖然核殼結構有良好的性能,但合成過程復雜,成本較高,可能比工業上使用的傳統催化劑昂貴得多。而且,合成此類結構涉及多個步驟,處理時間長,產量低,過程中使用多種化學品,產生較多的廢水和廢物,因此,核殼催化劑在大規模應用中的經濟吸引力遠遠低于當今的工業催化劑。研究可大規模生產、成本更低的核殼結構催化劑也非常重要。
猜你喜歡
哲學評論(2021年2期)2021-08-22 01:53:34
中華詩詞(2019年7期)2019-11-25 01:43:04
模具制造(2019年3期)2019-06-06 02:10:54
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
影視與戲劇評論(2016年0期)2016-11-23 05:26:01
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
現代企業(2015年9期)2015-02-28 18:56:50
