沉淀劑對NiW/TiO2-ASA催化劑加氫裂化性能的影響
2023-03-01 07:40:12孔倩孫巾超葛佳琪張鵬馬艷龍劉百軍
化工進展 2023年1期
關鍵詞:催化劑
孔倩,孫巾超,2,葛佳琪,張鵬,馬艷龍,劉百軍
(1 中國石油大學(北京)重質油國家重點實驗室,北京102249;2 國能朗新明環保科技有限公司,北京100039;3 中國石油石油化工研究院,北京102206)
無定形硅鋁具有豐富的介孔,被廣泛用于石油煉制催化劑,特別是作為加氫裂化和加氫精制催化劑的載體。通過引入第三金屬氧化物如TiO2等可調節無定形硅鋁的酸性質和織構性質。目前,研究者已嘗試使用多種方法制備TiO2?ASA,其中包括:徐培力等[1]使用硫酸鋁法制備無定形硅鋁干膠,以偏鋁酸鈉、硫酸鋁、硅酸鈉分別為鋁源和硅源,并流進料,后加入偏鈦酸溶液制得,所得含鈦無定形硅鋁干膠比表面積在350~460m2/g,孔容在0.7~1.3mL/g。萬國賦等[2?4]使用擬薄水鋁石、正硅酸乙酯、鈦醇鹽,采用溶膠?凝膠法制備Al2O3?SiO2?TiO2,以其為載體所得催化劑的比表面積在200~300m2/g之間,但孔容、孔徑較小,孔容在0.3~0.4mL/g,孔徑在6nm 左右。顧永和等[5]研究了原位溶膠凝膠法制備TiO2?SiO2?Al2O3三元載體,采用鈦、硅的醇鹽制備復合氧化物的前體,以無水乙醇為溶劑,使用十六烷基三甲基溴化銨為模板劑,所得三元復合載體的比表面積在200~300m2/g之間,孔容在0.6mL/g左右。周亞松等[6]采用溶膠?凝膠法制得鈦硅復合介孔材料,再使用同步或分步法向其中引入Al2O3,使用原位生長法將氧化鋁引入鈦硅氧化物中,所得載體比表面積在200~310m2/g,孔容為0.5~1.2mL/g。目前,合成高比表面積和大孔容鈦改性無定形硅鋁的方法多為溶膠?凝膠法,采用有機原料,且引入模板劑,成本較高,周期較長,工藝較復雜,實際所得的鈦改性無定形硅鋁多為高硅鋁比條件下制得,且比表面積和孔容仍比較小。另外,加氫裂化催化劑的加氫組分一般選擇ⅥB族和Ⅷ族元素的硫化或氧化物,研究發現,Ni和W組合的催化劑活性更好[7],故選用Ni、W 作為催化劑的加氫組分。現有研究多考察無定形硅鋁與分子篩復合作為載體的催化劑的加氫裂化性能,對于無定形硅鋁基復合氧化物作為單一載體組分的考察很少,為探究無定形TiO2?ASA 的加氫裂化性能,本研究以TiO2?ASA 為單一載體組分進行考察。
本研究以合成高比表面積、大孔容的TiO2改性無定形硅鋁為目標,采用分步沉淀法,在硅鋁比為1∶1的條件下,對比了4 種沉淀劑對TiO2?ASA 比表面積、孔容和酸量的影響。同時以TiO2?ASA為載體、Ni和W為加氫組分,制備NiW/TiO2?ASA催化劑,以十氫萘為反應的模型化合物,考察不同沉淀劑制備的NiW/TiO2?ASA催化劑在加氫裂化反應中的差異性。
1 實驗
1.1 TiO2-ASA的合成
將沉淀劑、硫酸鋁、硅酸鈉溶液(水玻璃)和硫酸氧鈦配成一定濃度的水溶液,備用。
首先在攪拌情況下并流加入硫酸鋁溶液和沉淀劑,控制反應體系的pH在8.0~9.0之間,同時控制反應體系的溫度在50~70℃,完成后老化60min。然后加入水玻璃溶液,完成后繼續在合成的溫度下老化60min。最后加入硫酸氧鈦溶液,完成后老化60min。將得到的產物經過濾、洗滌,120℃干燥8h、550℃焙燒3h,得到TiO2?ASA 樣品。合成過程中控制SiO2/Al2O3的質量比為1∶1,TiO2的質量分數為1%。使用的沉淀劑分別為氨水、碳酸銨、氫氧化鈉/尿素和氫氧化鈉,所得樣品依次記為TiO2?ASA?A、TiO2?ASA?B、TiO2?ASA?C、TiO2?ASA?D。
1.2 催化劑的制備
將一定量的TiO2?ASA與田菁粉、SB粉、稀硝酸按一定比例混合成團,擠條成形,經120℃干燥,550℃焙燒,得到催化劑載體。以硝酸鎳和偏鎢酸銨為Ni源和W源,使用等體積浸漬法,制備加氫裂化催化劑,活性金屬氧化物質量分數:WO3為24%、NiO為6%(以催化劑為基準)。將以TiO2?ASA?A、TiO2?ASA?B、TiO2?ASA?C、TiO2?ASA?D為載體制備的催化劑分別記為Cat?A、Cat?B、Cat?C、Cat?D。
1.3 樣品和催化劑的表征
采用德國Bruker公司生產的D8 Advance X射線衍射儀進行物相分析,Cu Kα射線,Ni濾波,管電壓40kV,管電流30mA。采用北京彼奧德電子技術有限公司KuboX 1000 全自動比表面積分析儀,進行樣品及催化劑的比表面積、孔結構測定,于350℃真空條件下預處理3.5h,之后通過氮氣吸附脫附進行測試。根據BET理論計算模型確定比表面積,孔徑分布依據Barrett?Joyner?Halenda(BJH)方法,孔容通過計算p/p0=0.98點的氮氣吸附量得到。采用天津先權公司TP?5079 全自動化吸附儀進行NH3?TPD表征,準確稱量0.100g樣品,于氦氣氣氛下升溫至400℃進行預處理,之后于室溫條件下進行NH3吸附,氦氣吹掃,程序升溫至600℃,記錄過程信號值。樣品和催化劑的吡啶紅外表征采用布魯克光譜儀器公司Bruker?80v 紅外光譜儀,取10mg 左右的樣品壓成圓形自撐片,于400℃進行預處理后,進行背景采集,吸附吡啶,分別記錄200℃和350℃處的脫附信號值,B酸、L酸的確定及酸量計算可參考文獻[8]。SEM 分析采用荷蘭FEI 公司Quanta 200F電子顯微鏡,樣品在測試前用物理方法鍍金。
1.4 加氫裂化反應評價
十氫萘的加氫裂化反應在中壓固定床微反裝置上進行。取5mL催化劑(20~40目)置于直徑10mm反應管中部恒溫段,首先進行預硫化,硫化液為3%CS2和97%環己烷(質量分數),壓力4.0MPa,體積空速2.0h-1,氫油體積比600m3/m3(標準狀況下),氫氣流量0.1L/min,硫化液流量10mL/h。加氫裂化反應條件為壓力4.0MPa,反應溫度360℃,空速2.0h-1,氫油體積比600m3/m3,產物經冷卻后收集,計算收集產物的質量。
以十氫萘的轉化率來衡量催化劑的裂化性能,以十氫萘的開環選擇性及產物收率來描述催化劑的加氫性能。計算如式(1)~式(3)。
式中,m1、m2、m3分別為單位時間十氫萘的進料量、液體產物質量、液體產物中開環產物的質量,g;w為液體產物中十氫萘的質量分數,%。反應產物用SP?3420氣相色譜儀(北京分析儀器廠)進行分析,SE?30毛細色譜柱(50m×0.25mm×0.5μm),氫火焰離子化(FID)檢測器。
2 結果與討論
2.1 XRD分析
不 同 沉 淀 劑 制 備 的TiO2?ASA 的XRD 譜 圖 如圖1 所示。由圖1 可知,4 種樣品均未出現明顯的TiO2特征衍射峰,說明使用不同沉淀劑制備的樣品中,TiO2以無定形或高分散的形式存在于復合氧化物中,在TiO2?ASA中,三種氧化物達到較高程度的混合,抑制了TiO2的晶型轉變。
圖1 TiO2?ASA粉體的的XRD譜圖
2.2 TiO2-ASA粉體的比表面積和孔結構
TiO2?ASA粉體的N2吸附?脫附等溫線如圖2所示,比表面積、孔容和平均孔徑的結果如表1。由圖2(a)可以看出,TiO2?ASA 粉體的吸附?脫附等溫線均為Ⅳ型,滯后環均為H4型,為兩端開孔的圓柱形細孔。從孔分布圖看,4種沉淀劑所制備的TiO2?ASA粉體的孔分布相差很大,氨水和碳酸銨為沉淀劑制備的TiO2?ASA 粉體孔分布比較集中,介于2~50nm之間,更多的是處于2~20nm之間。而以氫氧化鈉和氫氧化鈉/尿素為沉淀劑制備的TiO2?ASA粉體的孔分布比較彌散,尤其是以氫氧化鈉/尿素為沉淀劑制備的TiO2?ASA 粉體,它的孔分布是從2.2nm至100nm以上。

圖2 TiO2?ASA粉體的N2吸附?脫附等溫線和孔徑分布
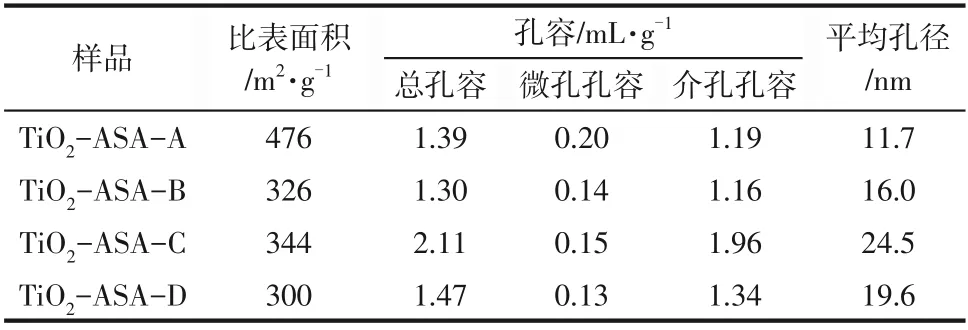
表1 TiO2-ASA粉體的織構性質
從表1的結果看,以氨水為沉淀劑的樣品比表面積最高,達到476m2/g,沉淀劑為氫氧化鈉時,樣品的比表面積最小,但都在300m2/g 以上,所制得的TiO2?ASA 都擁有較高的比表面積和較大的孔容,以此為載體制備的催化劑有利于擔載更多的活性金屬,有利于金屬粒子的分散及原料和產物的擴散。使用不同沉淀劑的過程差異對樣品的織構性質影響很大,氨水是弱堿,其產生OH-的速度適中,所以鋁離子充分成膠,給硅粒子與前體反應提供了更好的前提。氫氧化鈉作為強堿之一,可直接電離,因而鋁離子水解形成沉淀的速度較快,從而使TiO2?ASA比表面積較小。使用氫氧化鈉/尿素雙沉淀劑制備的樣品,在氫氧化鈉一種沉淀劑的基礎上,加入尿素,當溶液溫度高于60℃時,尿素水解生成OH-和CO2,尿素水解緩慢,有利于均相成核和生長[9],也有文獻[10]指出尿素可通過與鋁離子配位控制水解過程,在氫氧化鋁形成的初始階段廣泛成核。所以相對于使用單一氫氧化鈉為沉淀劑,其比表面積稍大。碳酸銨作為弱酸弱堿鹽,當產生的銨離子被不斷消耗,也就促進了其雙水解的進程,從而反應速度較快,所得TiO2?ASA比表面積較小。
所以,如果想要制備高比表面積、窄孔分布的TiO2?ASA,則以氨水和碳酸銨作為沉淀劑比較合適;如果追求的是大孔容、寬孔分布的TiO2?ASA,則以氫氧化鈉/尿素作為沉淀劑比較合適。
2.3 催化劑的比表面積和孔結構
NiW/TiO2?ASA 催化劑的N2吸附?脫附等溫線如圖3所示,比表面積、孔容和平均孔徑的結果如表2。從圖3可以看出,催化劑繼承了TiO2?ASA粉體的織構性質,吸附?脫附等溫線也都是Ⅳ型,滯后環為兩端開孔的圓柱形細孔的H4 型。值得一提的是,盡管TiO2?ASA粉體的比表面積相差較大,但催化劑的比表面積相差卻很小,都在200m2/g 左右,較大的比表面積有利于物質的吸附,為反應物提供更多的反應位點。催化劑的孔徑分布變化規律與TiO2?ASA 粉體也保持一致,即:氨水和碳酸銨為沉淀劑制備的催化劑孔分布比較集中,而以氫氧化鈉和氫氧化鈉/尿素為沉淀劑制備的催化劑的孔分布比較彌散。本研究以十氫萘為模型化合物,其分子直徑約為5.24nm,通過對催化劑的孔徑比較可知,所制得催化劑的最可幾孔徑均大于反應物分子直徑,所以反應物可以順利進入催化劑孔道發生反應。此外,如果制備大孔容、寬孔分布的催化劑,則選擇以氫氧化鈉/尿素、氫氧化鈉作為沉淀劑制備的TiO2?ASA粉體。
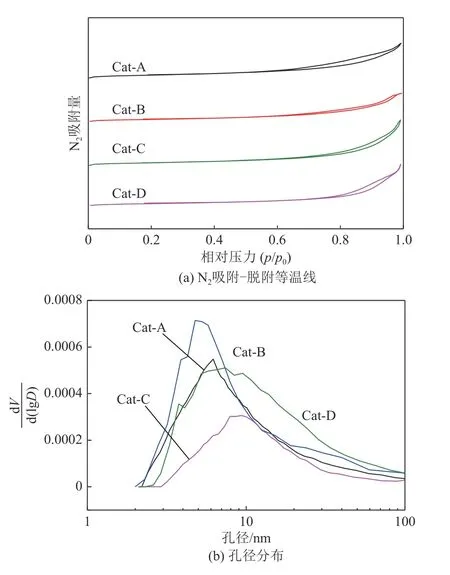
圖3 NiW/TiO2?ASA催化劑的N2吸附?脫附等溫線和孔徑分布
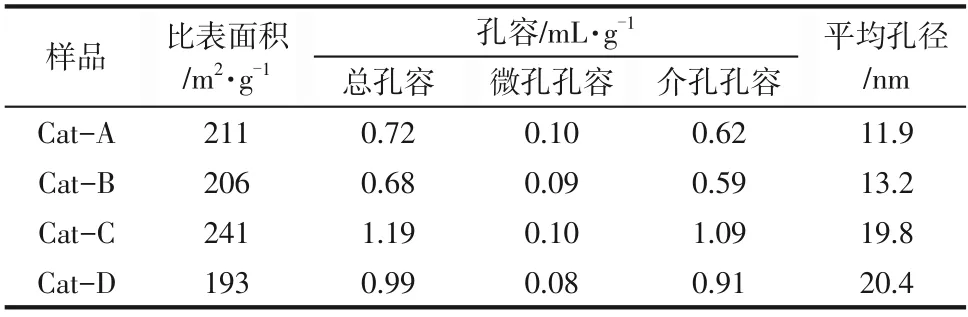
表2 NiW/TiO2-ASA催化劑的織構性質
2.4 催化劑的酸性表征
圖4 為不同沉淀劑制備的NiW/TiO2?ASA 催化劑NH3?TPD 曲線,4 種催化劑均出現較寬的脫附峰,多數對應弱酸位和中強酸位,當沉淀劑不同時,催化劑的酸性質相差較大。4條曲線的起點和終點基本重合,峰面積的差異明顯,SCat?A>SCat?B>SCat?C>SCat?D,所以Cat?A的總酸量最高,Cat?B次之,Cat?C、Cat?D 較小。觀察峰頂位置對應溫度,可以發現,Cat?B的脫附峰明顯向右偏移,向高溫方向偏移了大概15℃,此外,比較而言,Cat?B 的峰更“胖”一些,明顯“拖尾”,考慮中強酸的貢獻,因為ASA本身就屬于弱酸[11],中強酸含量較低,NH3?TPD對其響應度有限,使其弱酸及中強酸兩峰相互重疊,由此可見,Cat?B的中強酸更多,與表3所示吸附吡啶的紅外光譜結果基本一致。
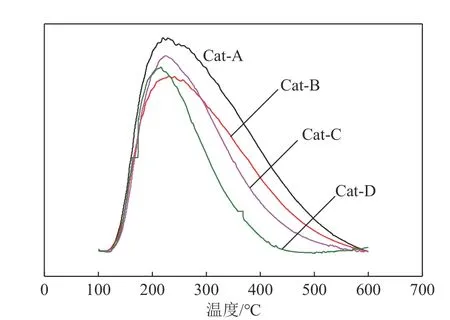
圖4 不同沉淀劑制備的NiW/TiO2?ASA催化劑NH3?TPD曲線
圖5和表3分別為NiW/TiO2?ASA催化劑的吡啶紅外光譜圖及根據紅外光譜計算得到的催化劑的酸分布結果,以氨水為沉淀劑時,催化劑具有最多的總酸量,以碳酸銨為沉淀劑總酸量次之,以氫氧化鈉為沉淀劑時,催化劑的總酸量最少,與NH3?TPD結果一致。沉淀劑為碳酸銨時,催化劑有最多中強酸的酸量和L酸的酸量。有文獻指出,Ti原子的加入有利于提高表面酸性,Al、Si、Ti原子間的的距離差異會導致表面酸度的不同,所以使用不同沉淀劑合成TiO2?ASA的過程差異,會直接影響酸性質[12]。
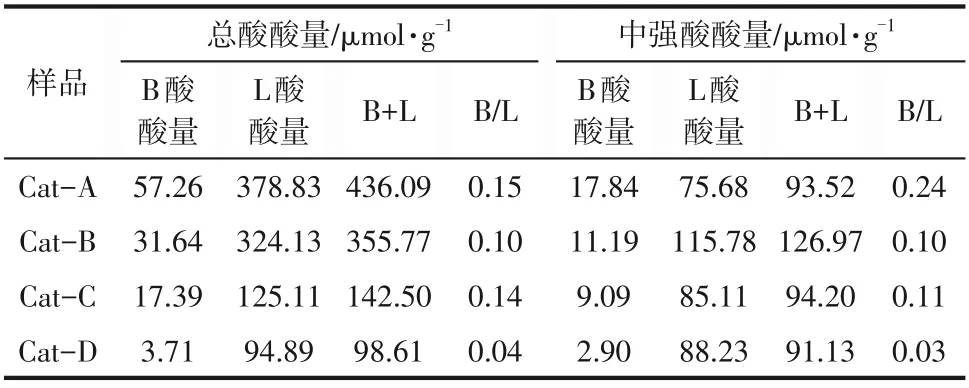
表3 NiW/TiO2-ASA催化劑的酸性分布

圖5 NiW/TiO2?ASA催化劑的吡啶紅外光譜圖
2.5 SEM表征
圖6 是不同沉淀劑制備的NiW/TiO2?ASA 催化劑的SEM圖。從圖中可以發現,使用不同沉淀劑制備的催化劑具有相似的微觀形貌,其狀態為無定形,均是由大小不一的不規則粒子堆積而成,并且每個粒子之間都形成了很多微米級孔隙,每個不規則粒子都是由許多更小的粒子堆積而成,這些小的粒子之間及內部也布滿了微小的孔道,以碳酸銨為沉淀劑時,催化劑的孔道結構較發達。以氫氧化鈉為沉淀劑時,組成顆粒的粒子變得更小,這些微小粒子堆積也變得更加緊密,其原因考慮與TiO2?ASA 樣品制備過程強堿的快速電離,鋁離子水解形成沉淀的速度過快有關。以碳酸銨為沉淀劑時,催化劑的孔道結構較發達,發達的孔道結構有利于反應物和產物的順利進出,有利于反應的順利進行。

圖6 NiW/TiO2?ASA催化劑的SEM照片
2.6 加氫裂化反應性能
表4 是十氫萘在NiW/TiO2?ASA 催化劑的加氫裂化性能評價結果。由表4可知,轉化率、開環選擇性、開環收率均顯示出同樣的規律,即:Cat?B最高,之后為Cat?A、Cat?C和Cat?D。
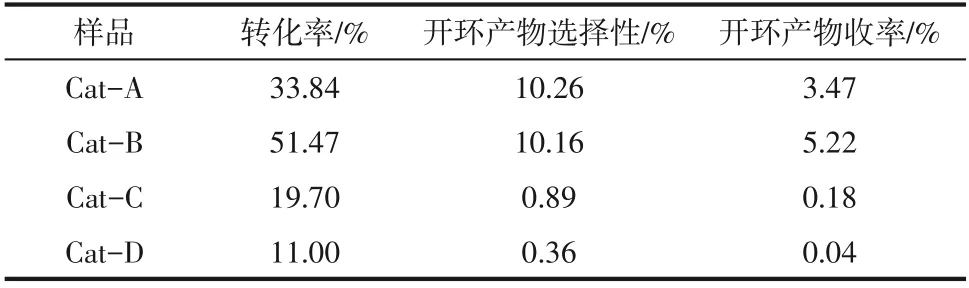
表4 NiW/TiO2-ASA催化劑的十氫萘加氫裂化性能
加氫裂化反應遵循正碳粒子反應機理,對于十氫萘,由于六元環本身具有最小的分子張力,是最穩定的環狀結構[13],因此對于類六元環類化合物,不可能直接開環得到其裂化產物,飽和環烷烴中的一個環從催化劑酸性位中得到一個質子,生成正碳離子化合物,六元環異構化為更穩定的結構,轉化為甲基環戊烷[14],再通過β?斷裂反應,使異構化產物裂化,轉化為更小的分子。開環作為其中的關鍵一環,因此把它作為反應的主要指標,在較低的轉化率下,比較催化劑的開環選擇性。反應開始于飽和環烷烴從酸性位得到質子生成正碳離子化合物,所以催化劑的酸性質對于加氫裂化反應至關重要。
無定形基催化劑中孔分布不均勻,酸性中心分布也不均勻。在低溫狀態下,主要起作用的是催化劑的強酸中心,對于催化劑的弱酸位而言,在溫度為360℃時也會表現出來,因此有利于異構化及裂化反應的進行[15]。對于反應物十氫萘而言,主要進行的就是異構化反應和裂化反應,在相同反應條件下,催化劑酸性越強,十氫萘的轉化率越高,催化劑酸性對十氫萘有重要影響[16]。本研究是以單一的無定形TiO2?ASA作為載體,沒有添加分子篩等其他強酸類材料,就TiO2?ASA本身而言,酸性較弱。因此,和添加分子篩的材料相比,催化劑加氫裂化反應性能較弱,對于TiO2?ASA 單一組分作為載體的催化劑而言,對比使用不同沉淀劑制備的催化劑,Cat?B具有較高的總酸量和中強酸量,所以其十氫萘的轉化率較高。有文獻提出,雙功能催化劑的載體對飽和雙環的開環反應影響很大,載體的結構和酸性分布會影響選擇性和產物分布,適量B酸與較多的L酸的存在,有利于脫氫十氫萘的質子化、氫原子的轉移、β鍵斷裂及開環過程[17?18]。有文獻指出,較低總酸量和較高L酸比例有利于提高中餾分的選擇性及保持較好的活性,L酸位與B酸位共存,會存在一定的協同效應,與B酸位鄰近的L酸位主要用于增強B酸位強度,因此在一定程度上增加了催化劑的催化活性,相比之下,Cat?C、Cat?D的總酸量和中強酸量都很少,尤其是Cat?D,其總B酸量為3.71,反應物從催化劑酸性位得質子生成正碳離子的過程就已經受阻了,所以其加氫裂化性能較差[19]。另外,對比使用4種不同沉淀劑制備的催化劑,其比表面積相差不大,Cat?A、Cat?B 的孔徑分布更集中,Cat?C、Cat?D的孔徑分布更彌散,與此同時,相較于Cat?A、Cat?B,Cat?C、Cat?D 的酸性相對較差,其反應活性也更差,說明酸性對于小分子加氫裂化反應是很重要的。Cat?B最可幾孔徑不大,酸性更好,催化活性更好,看似大孔徑更適合吸附、易于擴散,但對于使用十氫萘作為模型化合物來說,此孔徑范圍已可以滿足其吸附和擴散的基本要求,所以適宜的孔道結構更重要[20]。Cat?B 具有較大的比表面積、更合理的孔分布和酸分布,這些優良的織構性質和酸性質,是催化劑催化活性更好的主要原因,和前面的表征結果相符。
3 結論
(1)采用的沉淀劑不同,所制備的TiO2?ASA粉體的比表面積和孔結構差別很大,制備高比表面積、窄孔分布TiO2?ASA粉體,可使用氨水、碳酸銨為沉淀劑。制備大孔TiO2?ASA 粉體可使用氫氧化鈉/尿素為沉淀劑。
(2)不同沉淀劑制備的NiW/TiO2?ASA 催化劑的比表面積都能達到200m2/g 左右,相差很小,平均孔徑在10~20nm 之間,十氫萘的分子直徑約為5.24nm,使用4 種沉淀劑制備的NiW/TiO2?ASA 催化劑的最可幾孔徑均大于反應物分子直徑,反應物可以順利進入催化劑孔道發生反應。
(3)酸性對于小分子的加氫裂化反應很重要,使用氫氧化鈉/尿素、氫氧化鈉為沉淀劑時,盡管孔徑分布更彌散,但是其酸性相對較差,反應活性也較差,所以起決定作用的是催化劑的酸量,尤其是中強酸酸量,比表面積和孔結構的影響較小。以碳酸銨為沉淀劑制備的NiW/TiO2?ASA 催化劑具有最多的中強酸酸量,十氫萘的轉化率最高達到52%,開環產物選擇性達到了12%。
猜你喜歡
大自然探索(2023年7期)2023-11-14 13:08:06
石油石化綠色低碳(2019年6期)2019-02-13 09:39:01
石油石化綠色低碳(2019年6期)2019-01-14 01:16:22
智富時代(2018年3期)2018-06-11 16:10:44
浙江大學學報(工學版)(2016年11期)2016-06-05 09:21:04
Coco薇(2016年2期)2016-03-22 02:45:06
超硬材料工程(2016年1期)2016-02-28 22:20:04
中國資源綜合利用(2016年4期)2016-01-22 08:27:23
合成化學(2015年4期)2016-01-17 09:01:27
應用化工(2014年3期)2014-08-16 13:23:50
